The Perple_X
programs BUILD, VERTEX, FRENDLY, MEEMUM, WERAMI and PSSECT look for an option
file that contains keywords that control computational behavior. This option file is specified in the problem definition file
generated by BUILD and defaults to perplex_option.dat. If no option file is found,
then Perple_X uses the default values for each
option. This page
documents these options. The actual values for all relevant options are output
to the user console when a program is run. Perple_X
solution model and thermodynamic data files (e.g., solution_model.dat)
may also contain optional keywords that control how data is
treated. Some of these keywords are documented here.
Entries in
the option file take the general form
keyword value1 value2 ....
| commentary
where the keyword identifies an option and the associated values (e.g.,
on, off or a number) characterize the option. Comments may be placed
anywhere in the file provided they are preceded by a "|" character. In Perple_X
6.8.1+, any keyword may be assigned the value "default", in
which case the keyword is assigned its default attributes.
BEST
PRACTICE: to avoid confusion, create personalized and/or problem
specific option files (e.g., my_problem_option.dat) that list only those options
that have non-default values.
NOTE: Perple_X
PostScript plotting programs (e.g., PSSECT, PSVDRAW, PSPTS,
PSTABLE) read an additional option file. The
second option file, named
perplex_plot_option.dat, specifies plot options,
see
Perple_X
plot options for details.
The
6.9.1 version of Perple_X uses an iterative strategy in which the
continuous composition space of a solution phase is initially represented by a
static set of discrete compositions, i.e., pseudocompounds. Stable and nearly stable
compositions, i.e., refinement points, are identified from this representation
by linear programming (LP). The composition
space about the refinement points is then resampled and the LP optimization
repeated until a specified precision has been achieved. The resampling between
LP optimizations is done by quadratic programming (QP), which optimizes the composition of each refinement point relative to the
current LP solution. Phase diagram
sections are calculated by the program vertex
in two stages: an exploratory stage and an auto-refine
stage. During the exploratory stage a low resolution set of static
compositions is used to determine the approximate range of the stable phase
compositions in the section. The compositions determined in this manner form the
set of static compositions considered during the auto-refine stage. The details
of this algorithm and various thresholds are controlled by options that are
documented here collectively. Tuning with these options can be frustrating
because they are not independent and changing them may have non-intuitive
consequences.
Useful
options:
NOTE: setting intermediate_savrpc
and intermediate_savdyn
to T is the most general method of remedying
computational problems in vertex and meemum. When this method fails systematically explore the effect
of the options listed below. It is often helpful to use meemum
with the output_iteration_details
and output_iteration_G set to T
in order to understand these effects.
-
optimization_precision
- specifies the difference in Gibbs energy during successive LP
optimizations at which the optimization is considered to have converged;
default: 1e-4 J/mol; practical range: [1e2, 1e-6]. Small values increase
computational cost and may lead to failed optimizations (marked by red patches
in phase diagram sections); excessively large values cause rough phase
boundaries.
-
replicate_threshold
- during the exploratory stage of a calculation in vertex: a stable, or
nearly stable, composition is added to the list of static compositions to be
considered during the auto-refine stage of the calculation if it is separated
from all compositions in the list by a Euclidean distance greater than the
replicate_threshold value; default: 1e-3 mol; practical range: [1e-2,
1e-8].
replicate_threshold effectively defines the resolution of the static
compositional array used during the auto-refine stage. Small values
increase computational cost and may lead to failed optimizations (marked by
red patches in phase diagram sections); excessively large values cause rough phase
boundaries.
-
rep_dynamic_threshold
- during QP in vertex/meemum: if a dynamically-generated composition
is separated from a previous dynamically-generated composition by a Euclidean
distance smaller than the replicate_threshold value, then the composition is
not added to the list of compositions considered during subsequent LP
optimizations; default: 1e-3 mol; practical range: [1e-2, 1e-6]. Small values
increase computational cost and may lead to failed optimizations (marked by
red patches in phase diagram sections); excessively large values cause rough phase
boundaries.
-
intermediate_savrpc
- in vertex/meemum: controls whether compositions are added to the list of
dynamically generated compositions during intermediate stages of QP
optimization; default: F (false). Setting intermediate_savrpc to T (true) increases computational cost
and may increase optimization precision.
-
intermediate_savdyn
- during the exploratory stage of a calculation in vertex: controls whether compositions
from intermediate LP optimizations are added to the list of static compositions
to be considered during the auto-refine stage of the calculation; default: F
(false). Setting intermediate_savrpc to T
(true) increases computational cost and may increase optimization precision.
Useless
new options: these options control aspects of the 6.9.1+ algorithm,
the default choices are likely optimal.
-
optimization_max_it
- if the number of iterations during successive LP exceeds
optimization_max_it iteration is terminated regardless of whether the optimization_precision
value has been achieved; default: 40.
-
MINFRC_diff_increment
- the increment used for evaluation of finite differences during QP when
analytical derivatives are not available; default: 1e-7; practical
range [1e-2, 1e-8].
-
scatter-points
- controls whether additional compositions are generated about
a final QP result; default: T (true).
Setting scatter_points to F (false) decreases computational cost.
-
scatter-increment
- the compositional increment used to generate the
compositions requested if the scatter-points
option is T; default 1e-2 mol; practical range [1e-2, 1d-7].
-
keep_all_rpcs
- controls whether the list of dynamic compositions is reset after
each LP optimization within a successive series; default: T (true).
Setting keep_all_rpcs to F (false) decreases computational cost and
optimization precision.
-
re-refine
- causes the auto-refine data file to be updated during the auto-refine stage
of a calculation in vertex; default: F (false). The updated
auto-refine data can be used in repeated auto-refine stage calculations if the
auto_refine keyword has been set to manual.
Some
observations re tuning relative to the default values of the
above options:
-
Setting intermediate_savdyn
to T, followed by setting intermediate_savrpc to
T, are the changes most likely to remedy rough phase boundaries.
-
Sequentially
increasing the values of the optimiziation_precision,
rep_dynamic_threshold, and replicate_threshold are the changes most likely to
remedy sections with irregular red patches indicative of failed
optimizations; if these values excessive, then computed phase boundaries
will roughen.
-
Setting intermediate_savdyn
to T is the change most likely remedy smooth red patches that may appear
in sections computed with lagged
aqueous speciation .
NOTE: abbreviation is a not a perplex_option
file
keyword, the abbreviation keyword is an option that applies to a specific
solution model. To implement this keyword modify the solution model by
adding the keyword and its value on a line after the solution model name.
The
abbreviation keyword specifies the abbreviation used by Perple_X
on output if solution_names
is set to abb[reviation]. The abbreviation must be < 7 characters, it
need not be unique to the solution model.
This keyword is only
relevant for WERAMI, PSSECT, and MEEMUM.
Other keywords relevant to solution model names: solution_names,
full_name.
The
absolute keyword controls whether WERAMI (computational modes 2-4)
outputs relative or absolute molar/mass compositions. Whether WERAMI outputs
molar or mass quantities is determined by the composition_phase
and composition_system
keywords.
This keyword may take values: T (true) or F (false). The default is
F.
The
Anderson-Gruneisen keyword controls the parameterization of high pressure polythermal equations of state (Murnaghan, Birch-Murnaghan).
This keyword may take values: T (true) or F (false). The default is
T.
If
the keyword is T [true], then the temperature derivative of the bulk
modulus is computed as a function of the pressure derivative of the bulk modulus
and the expansivity at the reference pressure as described by Helffrich &
Connolly (2009). If the keyword is F, then
the temperature derivative of the bulk modulus is assumed to be an independent
model parameter.
The utility of this option it corrects a tendency of certain thermodynamic
formulations (notably Holland & Powell 1998) to generate unrealistic physical
properties. This correction is particularly important for seismic velocity
calculations, but has the unfortunate consequence that it may influence
calculated phase equilibria. A means of implementing this correction for
purposes of seismic velocity calculations without affecting computed phase
relations is to set
Anderson-Gruneisen to
F for the calculation of phase equilibria in VERTEX
and subsequently set
Anderson-Gruneisen to T for the calculation of seismic velocities with
WERAMI. This method is not thermodynamically consistent, but the inconsistency
is unlikely to be of significance.
Other keywords relevant to seismic velocity calculations: vrh/hs_weighting,
bounds,
explicit_bulk_modulus,
poisson_ratio,
melt_is_fluid,
interpolation,
seismic_output, seimsic_data_file.
The
approx_alpha keyword controls whether the approximation exp(x)-> 1+x is
used to evaluate volume at the reference pressure for phases described by EoS
choices (1-4).
This keyword may take values: T (true) or F (false). The default is
T.
If
the keyword is T [true], then the approximation
V(T,Pr) ≈ V(Tr,Pr)·[1
+ integral(α(T,Pr),T=Tr..T)]
is
used, otherwise the exact form
V(T,Pr) = V(Tr,Pr)·exp[integral(α(T,Pr),T=Tr..T)]
is
used. Although the approximate form is inaccurate at high temperature it has
been assumed during the derivation of certain data bases (most notably that of
Holland & Powell 1998), therefore the approximation should be used with such
data bases to ensure consistency.
The aq_bad_results keyword
controls error handling during lagged
aqueous speciation calculations, it may take the following values:
err
- any suspect result is reported as a failed optimization.
101
- if a solute component is dissolved entirely in the solvent, cease iteration
and output the result. The results satisfy mass and charge balance, but
generally have not been refined to the target resolution (final_resolution)
and may reflect the influence of numerical instability.
102
- do not allow extrapolation of HKF into vapor-like density field or
coexistence of pure and impure solvent.
103
- implemented?
ign
- output all results regardless of suspect behavior.
The
default value is err for all computational modes other than 0-d phase
fractionation. The default for 0-d phase fractionation is 101.
See also: aq_solvent_solvus
The
aq_ion_H+ keyword controls whether the hydronium ion is used as the
reference electrolytic species (Eqs 3-8, Connolly
& Galvez 2018) in lagged
and simple back-calculated aqueous speciation calculations. The hydroxyl ion
is used as the reference species if aq_ion_H+ is false.
This keyword may take values: T (true) or F (false). The default is
T.
In lagged
aqueous speciation calculations, the chemical potentials from the previous
iteration of an adaptive optimization are used to compute the speciation of
electrolytic fluids. The aq_lagged_iterations keyword permits the user to
require additional intermediate iterations so that the lagged chemical
potentials converge on the current chemical potentials. The need for these
iterations can be evaluated by comparing the lagged aqueous speciation with the
back-calculated result (obtained if aq_output
is true). If the lagged and back-calculated results differ, then specifying a
small number of intermediate iterations will cause the results to converge. In
general intermediate iterations are unnecessary because they do not improve
numerical stability and because chemical potentials converge rapidly during
adaptive optimization (the chemical potentials can be monitored by activating output_iteration_G)
The
lagged_aq_iterations keyword takes an integer value, the default is 0.
See also: aq_output,
aq_solvent_composition,
aq_solute_composition,
output_iteration_G, species_output
The
aq_lagged_speciation keyword controls whether lagged
aqueous speciation calculations are done by WERAMI and MEEMUM. The
speciation is output only if a recognized solvent phase is stable and data for
aqueous species is present in the thermodynamic data file used for the
calculation.
This keyword may take values: T (true) or F (false). The default is
T.
See also: aq_output,
aq_solvent_composition,
aq_solute_composition,
output_iteration_G, species_output
The
aq_output keyword controls whether simple
back-calculated
aqueous speciation calculations are done by WERAMI and MEEMUM. The
speciation is output only if a recognized solvent phase is stable and data for
aqueous species is present in the thermodynamic data file used for the
calculation.
This keyword may take values: T (true) or F (false). The default is
T.
See also: aq_species,
aq_solvent_composition,
aq_solute_composition,
aq_lagged_speciation
The
aq_oxides keyword permits suppression of tests for chemical degeneracy
during lagged- and back-calculated speciation calculations. These tests must be
suppressed for such calculations if a thermodynamic data file specifying oxide
components is used (this practice is NOT
recommended).
This keyword may take values: T (true) or F (false). The default is
F.
See also: aq_species,
aq_solvent_composition,
aq_solute_composition,
aq_lagged_speciation
The
aq_species keyword controls the number of aqueous species, ranked by
abundance, output for simple back-calculated
aqueous speciation calculations done by MEEMUM and WERAMI (computational
mode 1). All aqueous species are output, without ranking, in WERAMI mode 2-4
calculations.
This keyword takes positive integer values. The default is 20.
The
aq_solute_composition keyword controls whether the concentrations of
solute species in simple back-calculated
aqueous speciation calculations are output in molal units (mole/kg-solvent)
or as mole fractions.
This keyword may take values: m (molar) or y (mole fractions). The default is
m.
The
aq_solvent_composition keyword controls whether the concentrations of
solvent species in simple back-calculated
aqueous speciation calculations are output in molal units (mole/kg-solvent)
or as mole fractions.
This keyword may take values: m (molar) or y (mole fractions). The default is
y.
The
aq_solvent_solvus keyword (lopt(46)) controls whether solute-free solvent
compositions are generated during lagged speciation calculations. This is
necessary if a vapor-like phase may separate from a solute-bearing fluid during lagged
speciation calculations. The current implementation of aq_bad_results may
interfere in phase separation problems and should be checked.
This keyword may take values: T (true) or F (false). The default is
F.
The
aq_vapor_epsilon keyword sets a threshold for the solvent dielectic
constant below which the solvent is considered to be vapor and incapable of
dissolving solutes. The current implementation of aq_bad_results may interfere
with the output of vapor-phase results and should be checked.
This
keyword takes a real value. The default is unity.
The auto_exclude
keyword prevents the inclusion of thermodynamic phases and species (typically
gas phase species) with no associated mechanical equation of state in Gibbs
energy minimization calculations. For instructions on how to associate a
molecular fluid EoS with gas species data see special_EoS.
This keyword may take values: T (true) or F (false). The default is
F.
The auto_refine
keyword controls whether Perple_X
(specifically the programs VERTEX, MEEMUM, and CONVEX) uses results from an "exploratory"
calculation to refine a subsequent recalculation of the same problem, i.e., an "auto-refined" calculation. The exploratory calculation is used to establish
the stable solution compositions.
This information is saved (even if auto_refine
is off) in a file named by concatenating the project name with the "arf"
file type suffix. For example, if the project
is named in, VERTEX or CONVEX generates a file named in.arf and,
if the option_list_files keyword is T, a
more easily interpreted version named in_auto_refine.txt.
In the auto-refine
stage of a calculation, VERTEX and CONVEX:
Eliminate
solutions that were not stable in the exploratory calculation.
Restrict
compositional ranges of solutions to the ranges established in the exploratory calculation.
Use
the second, auto-reine, value of the initial_resolution
and final_resolution keywords.
Change mapping
parameters for gridded minimization and phase
fractionation calculations or change variance and
search increment specifications for Schreinemakers and
mixed-variable diagram calculations.
MEEMUM
does not generate auto_refine data, but if such data exists from a calculation
with VERTEX or CONVEX, then MEEMUM prompts if the data should be used as above.
auto_refine
may be off, man, or aut; the default is man. If
auto_refine = man, VERTEX must be run independently for the exploratory and
auto-refine stages of a calculation. This manual mode allows users to
examine results from the exploratory stage before undertaking the more time
intensive auto-refine stage. If auto_refine = aut, VERTEX automatically
does both the exploratory and auto-refine stages in a single run.
auto_refine_file (modified
6.9.1)
The auto_refine_file controls whether the range of solution compositions
discovered during the exploratory stage of a calculation are output to a text file. If
auto_refine_file = T, then the file is output and named
my_project_auto_refine.txt, where my_project is the project name.
This keyword may take values: T (true) or F (false). The default is
F (prior to 6.9.1: T).
This keyword affects VERTEX and CONVEX.
The bad_number keyword determines the value
assigned to missing data or bad numerical results
(usually caused by floating point errors). Depending on the physical property of
interest it may be useful to change this value so that bad results can be
distinguished during post-processing (e.g., if seismic velocities are of
interest, assigning a negative bad_number allows identification of erroneous
velocities).
As of Perple_X
revision 6.6.5.9, bad_number may be set to NaN (not-a-number). The
advantage of setting bad_number to NaN is that advanced plotting programs such
as MatLab and, to a certain extent, PyWerami (i.e., in contour mode) treat NaNs
correctly as missing data. For example, if phase
composition is contoured as a function of pressure and temperature with bad_number set to NaN,
then the data will be only contoured where the phase is stable.
In contrast, if bad_number is set to zero, zeroes will be reported for the
composition wherever the phase is not stable; the zero
values (usually) imply a range of composition that is much greater than the true
range and consequently the default contour interval chosen by a contouring
program will not resolve the data well. The Perple_X
contour plot program PSTABLE is not capable of treating missing data and
replaces NaNs with zeroes; thus there is no advantage to setting bad_number to
NaN unless the data is to be processed with MatLab or PyWerami.
This keyword takes numeric values or NaN (< 12 characters),
NaN is the default
value.
This keyword affects WERAMI,
FRENDLY and VERTEX.
Because the time scale for seismic
wave propagation is short compared to the time scales for thermal and chemical
equilibration, the elastic moduli (i.e., adiabatic bulk and shear moduli)
relevant for the calculation of seismic wave speeds are not identical to the
isostatic (Reuss) limit assumed for thermodynamic calculations*. In the absence
of detailed textural information, the moduli are estimated as the weighted
average of upper and lower bounds on the moduli. Two methods for the
estimation of these bounds are the Hashin-Shtrikman (HS) and Voigt-Reuss-Hill (VRH)
methods (Berryman,
AGU Handbook of Physical Constants, 1995). The value of the bounds
keyword (HS or VRH) determines method is used.
HS bounds are narrower than the VRH bounds and are theoretically justified;
thus, they are to be preferred. Despite this advantage, the VRH bound is more
widely used in geoscience, presumably because it is simpler to compute.
If bounds = VRH, the lower
bound Z- (i.e., Reuss limit) on the modulus Z of an
aggregate is the harmonic mean of the corresponding moduli of its p
constituent phases
Z- = 1/[sum(xi/Zi,i=1..p)],
[1]
where xi is the
volume fraction of phase i; and the upper bound Z+ (i.e.,
Voigt limit) is the arithmetic mean
Z+ = sum(xi*Zi,i=1..p).
[2]
If bounds = HS, the lower and
upper bounds Z-/+ are
Z-/+ = 1/[sum(xi/[Zi-Y-/+],i=1..p)].
[3]
In
Equation 3: for the adiabatic shear modulus μ
Y-/+ = 4/3 μmin/max,
[4]
where μmin and μmax
are, respectively, the minimum and maximum shear moduli of the constituent phases;
and for the
adiabatic bulk modulus K
Y-/+ = μmin/max([9Kmin/max+8μmin/max]/[Kmin/max+2μmin/max])/6.
[5]
In Equations 3-5, the lower bound Z-
is computed with Y-, Kmin and
μmin; whereas the upper bound Z+ is computed with
Y+, Kmax and μmax.
For either value of bounds, the
aggregate modulus is
Z = (1-χ)*Z- + χ*Z+,
[6]
where the weighting factor χ is
specified by the vrh/hs_weighting keyword.
By
default bounds = VRH.
This option affects WERAMI,
MEEMUM and FRENDLY. For calculations analyzed with WERAMI, changing this keyword
does not necessitate repeating the primary calculation with VERTEX.
Other keywords relevant to seismic velocity calculations: vrh/hs_weighting,
explicit_bulk_modulus,
poisson_ratio,
Anderson-Gruneisen,
melt_is_fluid,
interpolation,
seismic_output.
*Prior
to November 21, 2018; Perple_X
computed the shear modulus of a solution as the molar-weighted mean of its
endmember shear moduli, Weidner et al. (JGR, 1984) cite empirical evidence in
support of this assumption and, in the absence of a non-isostatic EoS, there is
no theoretical constraint on the relation between the shear moduli of a solution
and its endmembers. However, discounting excess volume effects, the bulk modulus
of a solution is
given by the volume-weighted Harmonic (Reuss) average of the bulk moduli of
its endmembers. Therefore, simplicity can be invoked as an argument to support
the contention that the shear modulus of an impure phase should be estimated by
the Reuss average as suggested by Stixrude & Lithgow-Bertelloni (GJI, 2005).
In recognition of this argument the Reuss average has been employed in Perple_X
for this purpose since November
21, 2018. Thus criticism voiced by Garber et al. (G3, 2019) about the use of
the Voigt average in Perple_X
is apt, but only relevant to earlier versions of Perple_X
and Connolly & Kerrick (2002, and Connolly 2005).
The closed_c_space keyword controls whether compositional variables for
gridded minimization calculations define a closed or open composition space. In
gridded minimization calculations as a function of k compositional
variables X(C1)...X(Ck), the
user specifies k+1 bulk compositions C0...Ck,
if closed_c_space = T, then the bulk composition of the system
C is computed as
C = sum{X(Ci)*Ci, i=1...k}
+ C0*[1-sum{X(Ci), i=1...k}],
whereas if closed_c_space = F, then
C = sum{X(Ci)*Ci, i=1...k}
+ C0.
This keyword may take values: T (true) or F (false). The default is
T.
This keyword is only affects VERTEX.
The composition_constant keyword controls whether the terms in user
defined phase compositions may include a constant.
This keyword may take values: T (true) or F (false). The default is
F.
This option only affects WERAMI.
See also: composition_phase, composition_system,
proportions
The composition_phase keyword controls whether phase compositions are output in
molar or mass units.
This keyword may take values: wt, mol. The default is mol.
This option only affects WERAMI.
See also: absolute, composition_constant, composition_system,
proportions
The composition_system keyword controls whether the amounts of a systems
components are output in
molar or mass units.
This keyword may take values: wt, mol. The default is wt.
This option only affects WERAMI.
See also: absolute, composition_constant, composition_phase,
proportions
The
console_messages
keyword controls whether messages to the user console are output (provided a
print file is requested) during Schreinemakers and mixed variable diagram
calculations.
This keyword may take values:
on, off. The default is on.
This keyword
affects VERTEX.
The
cumulative keyword controls whether WERAMI in computational mode 3 for
property choice 36 (all system or phase properties) outputs the cumulative
absolute composition of a phase or system. This option is only useful for
computing the total mass or molar amount of components removed from a system by
the fractionation of a phase. For this purpose, the absolute
keyword must also be set to true. This keyword only affects WERAMI property
choice 36 and will result in nonsensical results if used in WERAMI computational
mode 2.
Whether
WERAMI outputs molar or mass quantities is determined by the composition_phase
and composition_system
keywords.
This keyword may take values: T (true) or F (false). The default is
F.
If the cumulative_modes keyword is set to on, then cumulative
phase modes (by volume or weight as determined by the
proportions keyword) are output if the "modes of all phases" property is selected
in WERAMI. Given a list of phases, the cumulative mode of a phase is the
mode of the phase plus the modes of all the phases that precede it in the list.
This form of output is useful for constructing diagrams in which the variation
in the proportions of the phases are plotted as a function of a third variable,
e.g., pressure along a geotherm (see
Example #24).
This keyword may take values:
on, off. The default is off.
NOTE: As of 12/20/08 keyword is no longer
read from the computational option file, rather it is set interactively if the
"modes of all phases" property is selected in WERAMI.
See also: fancy_cumulative_modes
The
dependent_potentials keyword controls whether dependent chemical potentials
are output when chemographic data is output (provided a print file is requested)
during non-adaptive minimization calculations. In adaptive minimization
calculations the dependent potentials are output to my_project.plt and can be analyzed with WERAMI.
WARNING:
the VERTEX output file format depends on the dependent_potential keyword value,
consequently output generated by the VERTEX with the keyword set to off cannot
be read by WERAMI or PSSECT if the keyword is changed to on, and vice versa.
There is no check for the consistency of the keyword value between programs,
thus any inconsistency will cause a "hard" crash (I/O errors).
This keyword may take values:
on, off. The default is off.
The efficiency
keyword specifies the level of redundant testing done during Schreinemakers
diagram calculations.
This keyword takes
integer values 1->5, the default is 3. High values increase
computational efficiency, but also increase the probability that a [pseudo-]univariant
curve may be skipped or incompletely traced.
This keyword
affects VERTEX.
The adiabatic bulk modulus
KS(T,P) is an implicit function of the Gibbs energy
used to compute phase equilibria
(Equation 4 of Connolly &
Kerrick 2002). By default, Perple_X
exploits this dependence to compute
KS(T,P). This method has the virtue that the modulus is consistent
with the equation of state used to compute phase relations. The importance of
this consistency for seismic velocity calculations is debatable because phase
relations depend directly on the Gibbs energy, whereas
KS(T,P) is dependent primarily on second order
derivatives of the Gibbs energy. Thus, it can be argued that empirically calibrated expressions for
KS(T,P) are more accurate than the values obtained
from an EoS intended only for phase equilibrium calculations (e.g., Holland &
Powell 1998). To accommodate this case, the explicit_bulk_modulus keyword
permits the use of empirical expressions for
KS(T,P).
If explicit_bulk_modulus = T (true),
KS(T,P) of any pure phase or endmember is computed
from the expression
KS(T,P) = k0 + k1·(T-Tr)
+ k2·(P-Pr)
provided that the parameters {k0, k1, k2} are
specified for the phase in
the thermodynamic data file. If explicit_bulk_modulus = T (true),
KS(T,P) for a solution phase is computed as the
volumetrically weighted mean of the endmember properties provided the parameters
{k0, k1, k2}. Under any other circumstances
KS(T,P) is computed by differentiation of the Gibbs
energy function for the phase.
This keyword takes the character values T
or F. The default value is F.
This option affects WERAMI,
MEEMUM and FRENDLY. For calculations analyzed with WERAMI, changing this keyword
does not necessitate repeating the primary calculation done by VERTEX.
Other keywords relevant to seismic velocity calculations: vrh/hs_weighting,
bounds, poisson_ratio, Anderson-Gruneisen,
melt_is_fluid,
interpolation, seismic_output, seimsic_data_file.
The extrapolation keyword is only relevant for gridded minimization and
has no effect unless interpolation (see
below) is also on.
This keyword may take values: on, off. The default is on.
This option only affects WERAMI, extrapolation may cause floating point errors
in Perple_X 07.
Cumulative
modes generated by WERAMI in computational mode 3 for
property choice 25 (modes of all phases) are, by default, ordered so that if a
new phase appears along a path its cumulative mode corresponds to its real mode
(i.e., the cumulative mode on the new phase initiates from zero). In contrast,
if fancy_cumulative_modes is T and the appearance of a new
phase is coincident with the disappearance of a phase, then the
cumulative mode of the new phase is computed so that the new phase is located in
the same relative position as the phase that disappeared. The rationale for this
scheme is that the coincidence is only possible if the two phases are related by
a univariant reaction. Finite resolution, i.e., internodal spacing, may have the
consequence that more than one phase may appear or disappear between two
adjacent nodes (i.e., the nodes may span multiple phase boundaries), in such
cases the fancy_cumulative_modes option may lead to erratic results.
This keyword may take values: T (true) or F (false). The default is
F.
The
fd_expansion_factor keyword is the factor by which the finite difference
increment used to compute high order derivatives increases with order. These
derivatives are used in Perple_X to compute
physical properties during post-processing.
This
keyword takes a real value. The default value is 2.
If
the first order finite difference increment for a variable is dv1, then
the increment used to compute nth order finite difference increment dvn =
dv1 * fd_expansion_factor^(n-1).
See
finite_difference_p for related information.
This keyword affects WERAMI, FRENDLY, and MEEMUM.
The final_resolution keyword determines the (molar) precision with which
phase compositions are calculated during adaptive minimization (Connolly 2009).
Because resolution is discretized in Perple_X,
the effective final resolution of adaptive minimization calculations is
determined by finding the minimum number of iterations that yield precision
below final_resolution. The effective final resolution is computed as outlined
in the resolution factor keyword description. See Example
1 of the adaptive
minimization keyword group discussion for additional information on the
effect of this keyword.
The
final_resolution keyword takes two values between 0 and 1. The first value
applies during the exploratory stage of auto-refine calculations and the second
value applies in the final (auto-refine) stage of the calculation. If only one
value is present it is applied for both stages, if the keyword is absent the
default targets for the final resolution in the exploratory and auto-refine
stages are 1e-2 and 1e-3 (mol), respectively. If final_resolution > initial_resolution, then there is no iterative
refinement of phase compositions and the effective resolution is determined by
the initial_resolution keyword.
The
value of final_resolution does not affect memory consumption, but small values
may significantly increase computational time. The minimum value of is limited
by the conditions at which the requested resolution causes numerical instability
(i.e., warning ver058, failed optimizations). These conditions depend on many factors, but, as a
rule of thumb, on 32-bit machines final_resolution should not be << 10-4
mol.
This keyword affects VERTEX and MEEMUM.
See
also: resolution_factor, reach_increment,
initial_resolution, output_iteration_G,
adaptive
minimization keyword group.
The
finite_difference_p keyword controls the magnitude of the finite
difference increment on pressure for first order derivatives. These derivatives
are used in Perple_X to compute physical
properties during post-processing.
The
keyword takes two real values, threshold and fraction. The default
value for threshold is 10000 and that of fraction is 0.01.
If
the pressure for a calculation is greater than threshold then the
pressure increment dp1 = pressure * fraction, otherwise the pressure
increment is dp1 = threshold * fraction.
See
fd_expansion_factor for related information.
This keyword affects WERAMI, FRENDLY, and MEEMUM.
In
phase fractionation calculations, the fractionation_hi_limit and fractionation_lo_limit
keywords permit specification of fractionation thresholds such that: 1) the mass
fraction of a phase must exceed the fractionation_hi_limit before it is
fractionated; and 2) the residual mass fraction of the phase after fractionation
is fractionation_lo_limit. If fractionation_hi_limit ≤
fractionation_lo_limit, then VERTEX emulates Rayleigh fractionation,
i.e., any fractionated phase is removed if its amount exceeds zero leaving no
residual.
Each
keyword takes real values ≤ 1. The default for both keywords is
zero.
NOTE:
full_name is a not a perplex_option file keyword, the full_name keyword is an option that applies to a specific
solution model. To implement this keyword modify the solution model by
adding the full_name keyword and its value on a line after the solution model
name.
The
full_name keyword specifies the full name used by Perple_X
on output if solution_names
is set to ful[l]. The full_name must be < 23 characters, it need not be
unique to the solution model. The full_name tag is also used by BUILD to group
solution models, e.g., to have BUILD group all orthopyroxene models together
after the solution model prompt, all orthopyroxene models should be given the
same full_name, e.g., 'orthopyroxene'.
This keyword
affects BUILD, WERAMI, PSSECT, and MEEMUM.
Other keywords relevant to solution model names: solution_names,
abbreviation.
The
global_reach_increment specifies an incremental increase in the range over which
the compositional variables of solutions are allowed to vary between successive
iterations during adaptive minimization.
Experience
suggests that except for immiscible solution models there is no benefit to
increasing this range beyond the default (see resolution_factor,
eq [1]) and because such an increase is costly in terms of memory, it is
preferable to apply such increases only for specific solution models via the
solution model file reach_increment keyword. If
global_reach_increment > reach_increment, its value overrides reach_increment,
otherwise the value of reach_increment is retained.
See
reach_increment for further details.
This keyword affects VERTEX and MEEMUM.
See
also: final_resolution, reach_increment,
initial_resolution, solvus_tolerance,
adaptive
minimization keyword group.
For gridded minimization VERTEX uses a multilevel grid refinement strategy in
which phase relations are mapped on an initial grid of resolution x_nodes
* y_nodes. This grid is refined grid_levels - 1 times by
bisection (Connolly 2005). The x_nodes, y_nodes, grid_levels
parameters are set by keywords of the same name. Each keyword takes two
integer values. The first value is used for the exploratory stage of gridded
minimization calculations, while the second value is used for the
auto-refine stage. The effective resolution
of a multilevel grid is equivalent to a single grid with (x_nodes - 1) * 2**(grid_levels
- 1) + 1 in the x-direction and (y_nodes - 1) * 2**(grid_levels - 1) + 1 nodes
in the y-direction.
The danger of using low values for x_nodes and y_nodes and a high
number of grid_levels is that a
small phase field that protrudes into a larger phase field will not be
identified if the protrusion occurs between two nodes of the low resolution grid
at which the larger field is stable. There is no simple rule for the best
choices for x_nodes or y_nodes as they depend on the scale of the
diagram and the heterogeneity of its phase fields, the default values are 10 (prior
to 6.9.1: 40) and
40 for exploratory and auto-refinement stages of a calculation. The default
values of grid_levels are 1 and 4 for exploratory and auto-refinement
stages.
y_nodes is used for 1-d gridded minimization calculations.
For 1-d phase fractionation calculations the 1d_path keyword indicates
the number of nodes taken to define the path in the exploratory and auto-refine
stages of the calculation. The default values for 1d_path are 40 and 150 for the
exploratory and auto-refinement stages of a phase fractionation calculation. As
a rule of thumb, the latter value should be chosen such that the results do not
change significantly if it is varied by a factor of two.
This keyword affects VERTEX.
If the hard_limits keyword is set to off, then unnatural
composition limits (i.e., X < 1 or X > 0) specified in the solution model file
(e.g., solut_07.dat) may be relaxed during adaptive minimization calculations. This
behavior permits users to specify restricted compositional ranges for complex
(costly) models, yet allows VERTEX to learn new limits in the course of a
calculation. hard_limits should be set to on if a solution model
is duplicated in the solution model file in order to represent distinct
compositional ranges of the solution. For example, if a model for Na-K mica is
duplicated to distinguish Na- and K-rich micas. In this case it is conceivable
that if calculations were done under conditions where the mica is fully
miscible, VERTEX might expand the composition limit of Na-rich mica into the
range of K-rich mica, or vice versa, resulting in confusing output.
This keyword may take values:
on, off. The default is off.
This keyword affects VERTEX and MEEMUM.
The
hybrid_EoS_XXX keyword, where XXX may be H2O, CO2,
or CH4, determines the internal molecular fluid equation of state to be
associated with a pure XXX species fluid. The keyword may take integer values:
0 - MRK, Modified Redlich-Kwong EoS, DeSantis et al. 1974
1 - HSMRK, Kerrick & Jacobs 1981, invalid at P > ~4 GPa and at ambient
conditions.
2 - CORK, Holland & Powell 1998, valid only for H2O and CO2, invalid at
ambient conditions.
3 - BRMRK, Bottinga & Richet 1981, valid only for CO2
4 - PSEoS, PSEoS Pitzer & Sterner 1994, valid only for H2O and CO2, invalid
at ambient conditions.
5 - Steam Table EoS, Haar et al 1982, valid only for H2O
6 - Zhang & Duan 2005, valid only for water, invalid at T < ~700 K.
7 - Zhang & Duan 2009, invalid, at least for H2O, at T < ~700 K; may be
preferable to the MRK (current default) for CH4.
The
default values for hybrid_EoS_H2O, hybrid_EoS_CO2, and hybrid_EoS_CH4
are 4, 4, and 0, respectively.
The increment keyword specifies the default search/trace variable increments for
Schreinemakers and mixed-variable diagram calculations. Two values are
specified, these are applied, respectively, for the exploratory and autorefine
stages of calculation. The values of increment are relative, absolute increments
are computed by mutliplying the increment value by the variable range.
This keyword affects VERTEX.
VERTEX and MEEMUM, 6.9.1+:
VERTEX
and MEEMUM read only one value for the initial_resolution keyword (if two
values are present, the first value is applied). This value determines the default (molar) compositional
resolution used for solution models in the exploratory stage of calculations with
VERTEX. The value is also used in MEEMUM in calculations that do not use
auto-refine data from a previous calculation with VERTEX. If initial_resolution is greater than zero,
its value over-rides the resolutions specified for individual phases in the
solution model file.
initial_resolution takes a value between 0 and 1 and may be specified
either as an integer fraction (e.g., 1/2) or real number. If initial_resolution is zero,
then Perple_X will use the resolutions
specified in the solution model file. Optimally, initial_resolution
should be specified as an integer fraction of 1, other choices lead to
asymmetric resolution with a widow composition near one that can be costly in
calculations involving complex solution models. Experience suggests that a value
of 1/4 is adequate for most calculations.
CONVEX and 6.9.0-:
The initial_resolution keyword takes two values that determine the default (molar) compositional
resolution used for solution models in the exploratory and auto-refine
stages of calculations with VERTEX, CONVEX, and MEEMUM. If initial_resolution is greater than zero,
its value over-rides the resolutions specified for individual phases in the
solution model file. For the adaptive minimization calculations (MEEMUM and VERTEX) the effective initial resolution (d0) is
the appropriate, i.e., exploratory or auto-refine stage, value of
initial_resolution
and the final precision is specified by the final_resolution
keyword. For unconstrained minimization calculations (CONVEX), initial_resolution
specifies the effective compositional resolution.
In adaptive minimization calculations, the initial_resolution keyword controls
the amount of memory allocated to static pseudo-compounds. Small
initial_resolution values improves accuracy, but are costly in terms of both memory and computer time. The diagnostic **error ver041**
in calculations with VERTEX, CONVEX, and MEEMUM indicates that initial_resolution is
too small.
This keyword takes two values between 0 and 1 and that may be specified as integer
fractions (e.g., 1/16) or real number. If initial_resolution is zero,
then Perple_X will use the resolutions
specified in the solution model file. Optimally, initial_resolution
should be specified as an integer fraction of 1, other choices lead to
asymmetric resolution with a widow composition near one that can be costly in
calculations involving complex solution models. Experience suggests that the optimal values for initial_resolution
1/16 and 1/48 for most calculations and for composition diagram calculations in
CONVEX 1/16 and 1/160 (or less).
This keyword affects VERTEX, CONVEX, and MEEMUM.
The
interim_results keyword controls whether interim results are output by
VERTEX during 2-dimensional multilevel gridded-minimization calculations. During
each successive level roughly doubles the required computational time. Setting interim_results
to auto or manual causes VERTEX to output data which can be
plotted or analyzed with PSSECT and WERAMI before VERTEX has completed a
calculation. This option allows the user to identify computational problems
before the final result has been generated.
This keyword may take values:
off, auto, manual. The default is auto.
If
interim_results is set to auto, then PSSECT/WERAMI automatically
select the latest interim result for analysis and VERTEX automatically deletes
all interim results upon completing a calculation.
If
interim_results is set to manual, then PSSECT/WERAMI allow the
user to select from among interim results generated by VERTEX and VERTEX does
not delete the interim results upon completing a calculation.
If
interim_results is set to off, then no interim results are output
by VERTEX.
This keyword affects VERTEX, WERAMI, and PSSECT.
The interpolation
keyword is only relevant for gridded minimization and is currently only used for
mode 2 calculations (sampling on a grid) in WERAMI.
WARNING: turning interpolation "on" slows data processing.
The interpolation
keyword takes 2 values, value1 and value2
value1 is on
or off, the default is on
value2 is an
integer, the default is 2
When value1 is
on WERAMI
computes physical properties by interpolation/extrapolation of the properties of
an assemblage at a sampling point by linear interpolation and/or extrapolation
from the 3 closest grid nodes of the computational grid at which this assemblage is known to be
stable. For this purpose WERAMI uses an algorithm that tries
to locate 3 nodes bounding the sampling point, when this not possible
extrapolation is used if the extrapolation keyword value is on.
Figure 1 illustrates some consequences of interpolation
and/or extrapolation. The primary danger of interpolation as currently
implemented (i.e., this will change) is that it may homogenize nodes that
represent 2 or more compositions of a phase separated by a solvus.
Value2
specifies the the range of nodes around the sampling point searched with the
triangulation algorithm. For a multilevel grid with JLEV levels, the number of
nodes on around the sampling point is value2*(2**(JLEV-1)).
Figure 2
illustrates the effect of increasing value2, value2 = 0 is
equivalent to turning interpolation off, small values increase the probability
that WERAMI cannot find bounding interpolation points (in which case no
interpolation done around the sample point), whereas large values reduce the
accuracy of linear interpolation.
Figure 1. Water
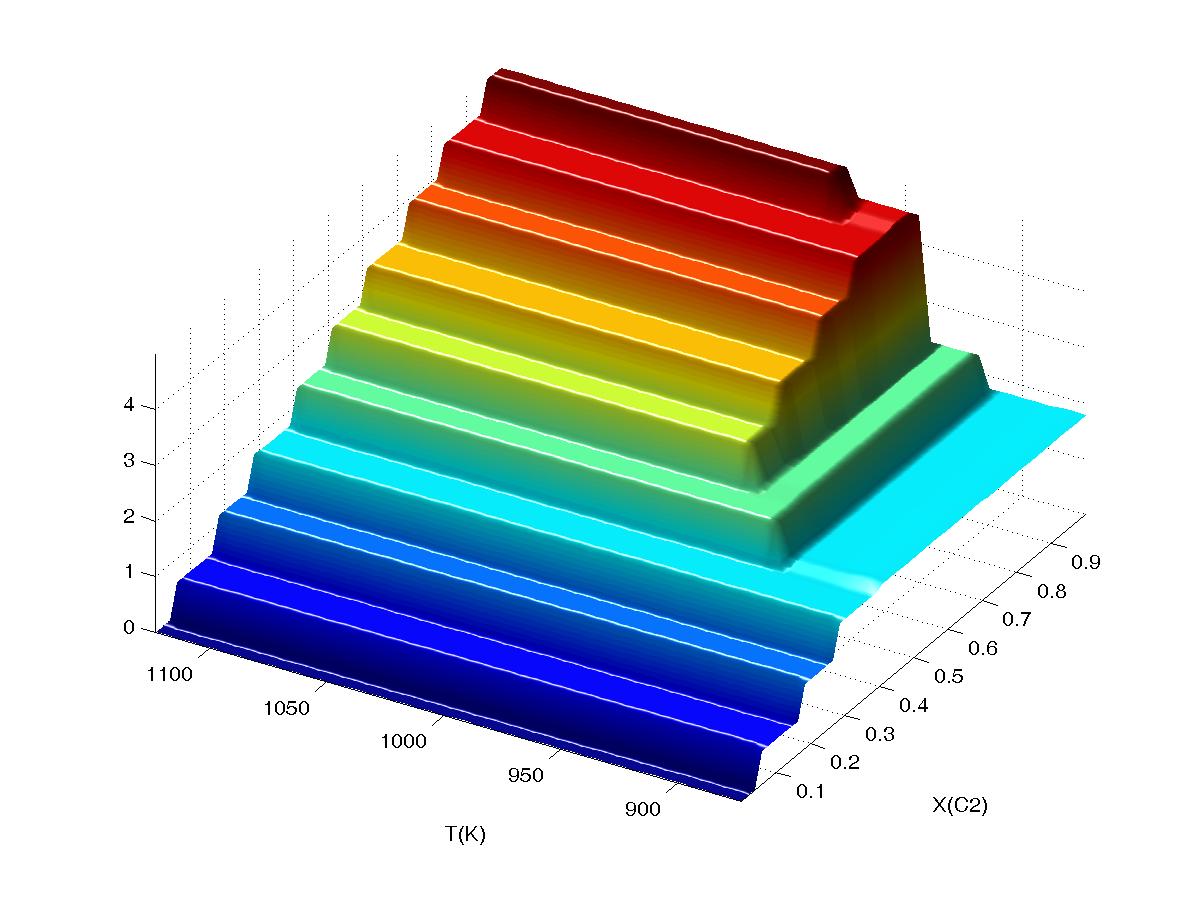
content of mineral+melt (i.e., excluding free water) assemblages for a calculation as a function of temperature
(T) and the compositional variable X(C2) which represents the bulk water content
of the system (from 0 to 5 wt%). The calculation was done on a low resolution
(single level 20x20) grid, for plotting purposes this grid was sampled on a
50x50 grid with WERAMI. At high temperature all H2O component is dissolved in
the melt phase and therefore the water content of the mineral+melt assemblage should vary linearly from 0
to 5 wt % as a function of X(C2), this is not the case if interpolation is
turned off (first plot, "interpolation off") because the water content of
the system is identified with discrete nodes of the grid. The advantage of
viewing the data with interpolation off is that it provides an unambiguous
picture of the raw data. Continued...
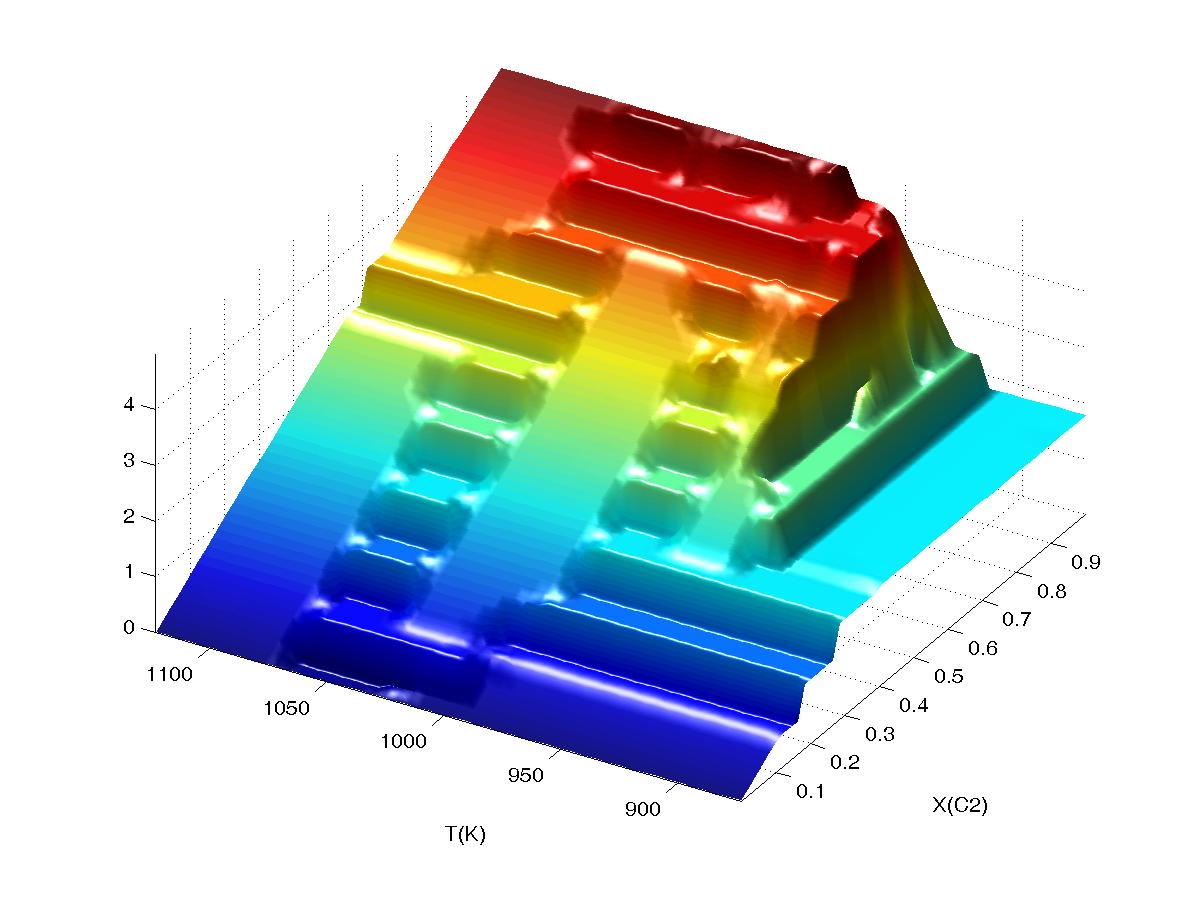
If interpolation is turned on, and
extrapolation turned off (second plot, "interpolation on 2" and "extrapolation
off"), the plot is smoothed, but discontinuities persist where WERAMI was unable to obtain identify three grid points representing the phase
assemblage bounding the sampling points. Continued...
If both interpolation and
extrapolation are on (third plot, "interpolation on 2" and "extrapolation
on") the required smooth variation in water content at low temperature is
obtained, although artificial plateaus persist where a particular phase
assemblage is stable only at one node (e.g. at T~1050 K, X(C2)~0.6) or
along a one-dimensional path (e.g., T<950 K, X(C2)~0.2). A disadvantage of
allowing extrapolation, particularly in the case of sections with compositional
variables, is that physically unrealistic values can be obtained, as at T~950 K,
X(C2)~1 where the water content depicted is greater than the maximum possible
for the system. The undesirable artifacts of extrapolation and interpolation
depicted here become less significant and less frequent as resolution of the
grid is increased.
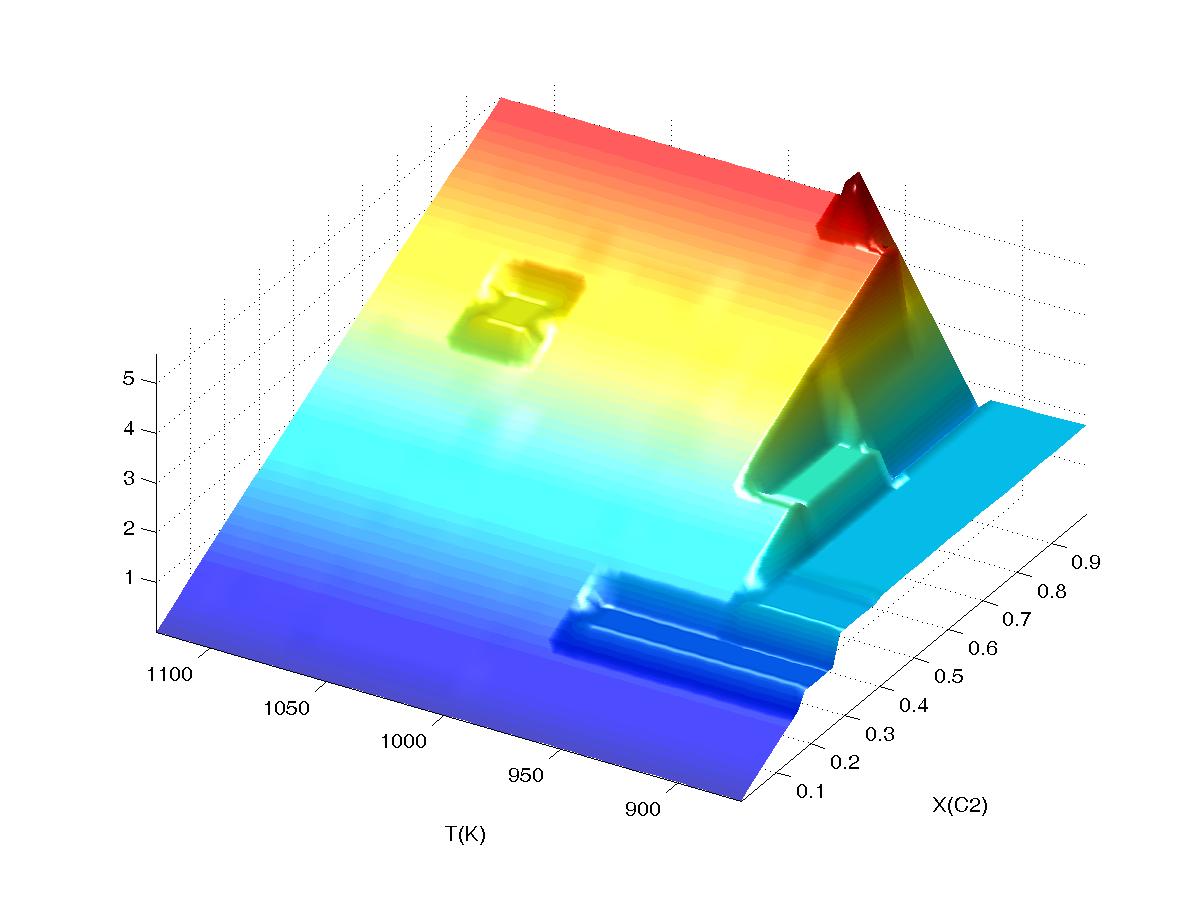
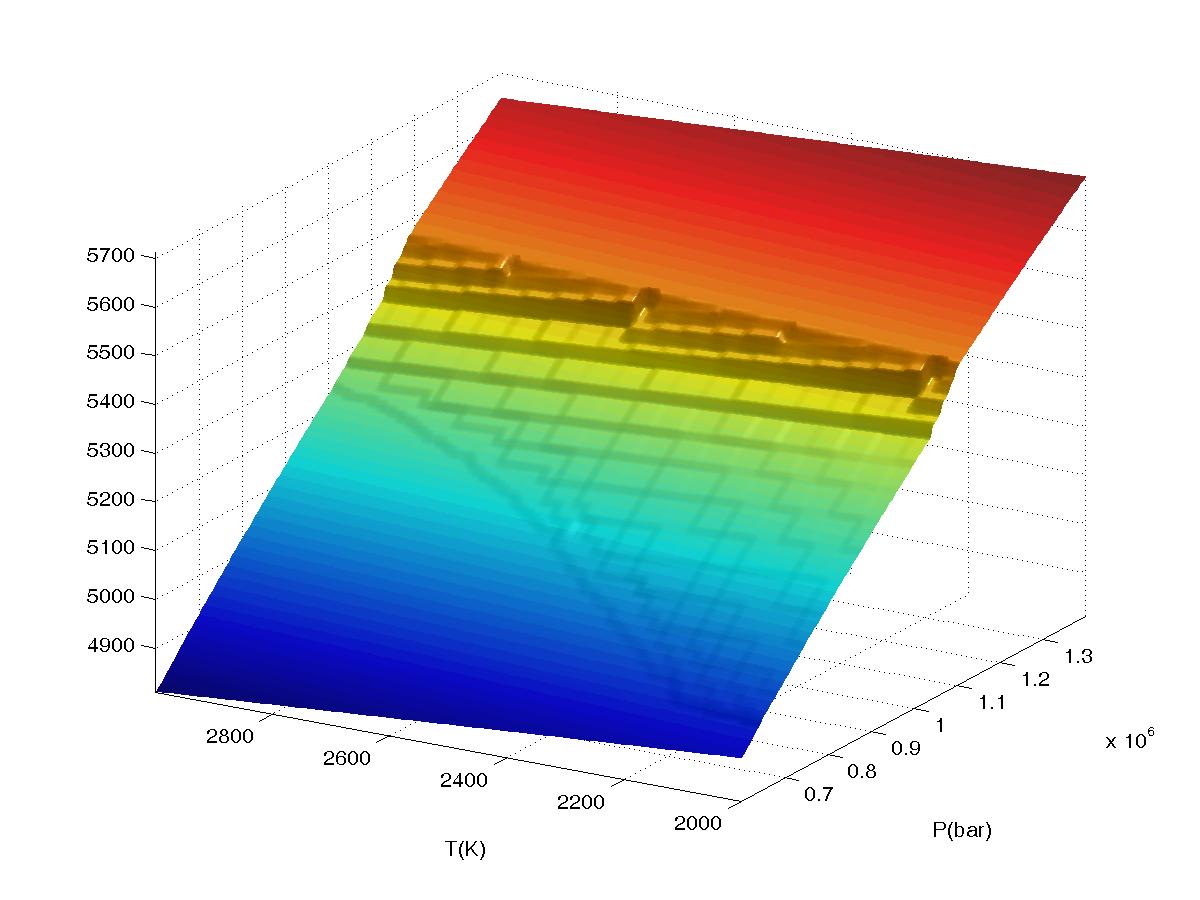
Figure 2.
Plots of bulk density for a high pressure phase transformation as a function of
interpolation range (value2), the calculation was done in VERTEX on a 4
level grid (JLEV=4) with 20x20 nodes at the lowest level of resolution and
refinement of only
true phase boundaries (the default grid refinement mode). The data
was sampled on a 150x150 grid in WERAMI to create the plots. All plots were made
with both interpolation and extrapolation on. The first plot shows results
obtained for value2=0 (equivalent to "interpolation off") in which the
underlying structure of the multilevel grid is clearly visible. Continued...
The second plot shows results obtained for value2=1, for JLEV=4 a unit
increment in value2 corresponds to 8 grid nodes at the highest level of the
computational grid. Continued...
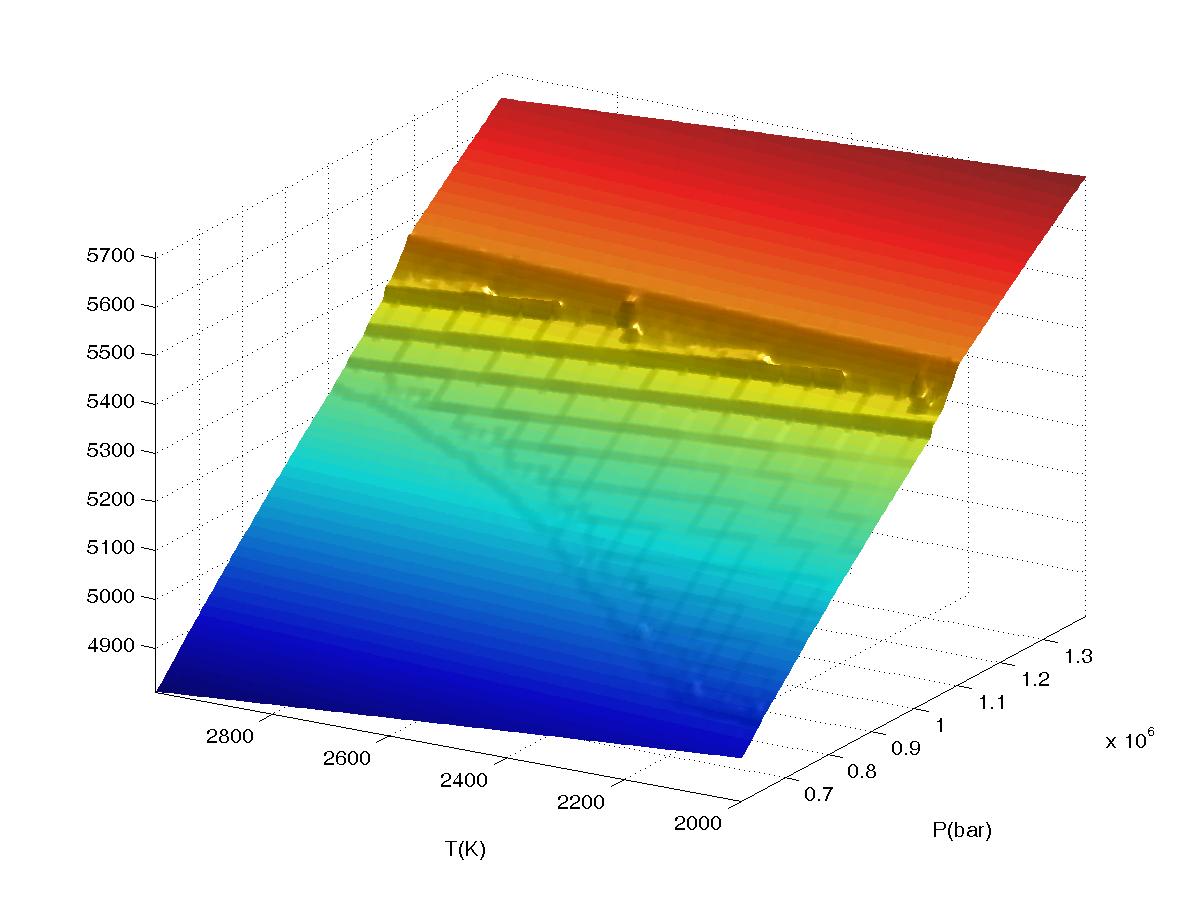
The third plot shows results obtained for value2=2 (the default value),
with this choice the continuous variation in density is reasonably well
represented. Although the surface appears eroded in places, probably a
result of extrapolation. Continued...
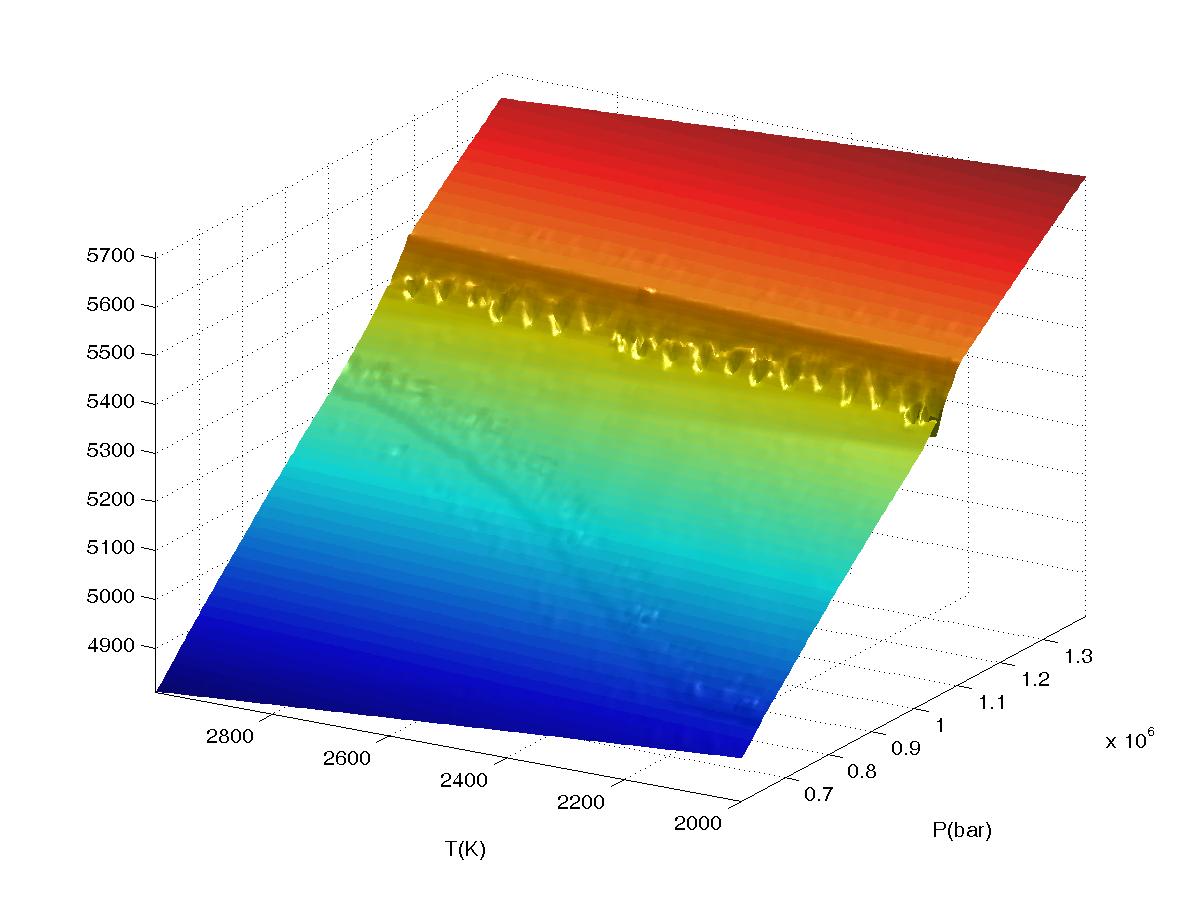
The fourth plot shows results obtained for value2=3, here the relatively
long interpolation/extrapolation distances cause undesirable artifacts.
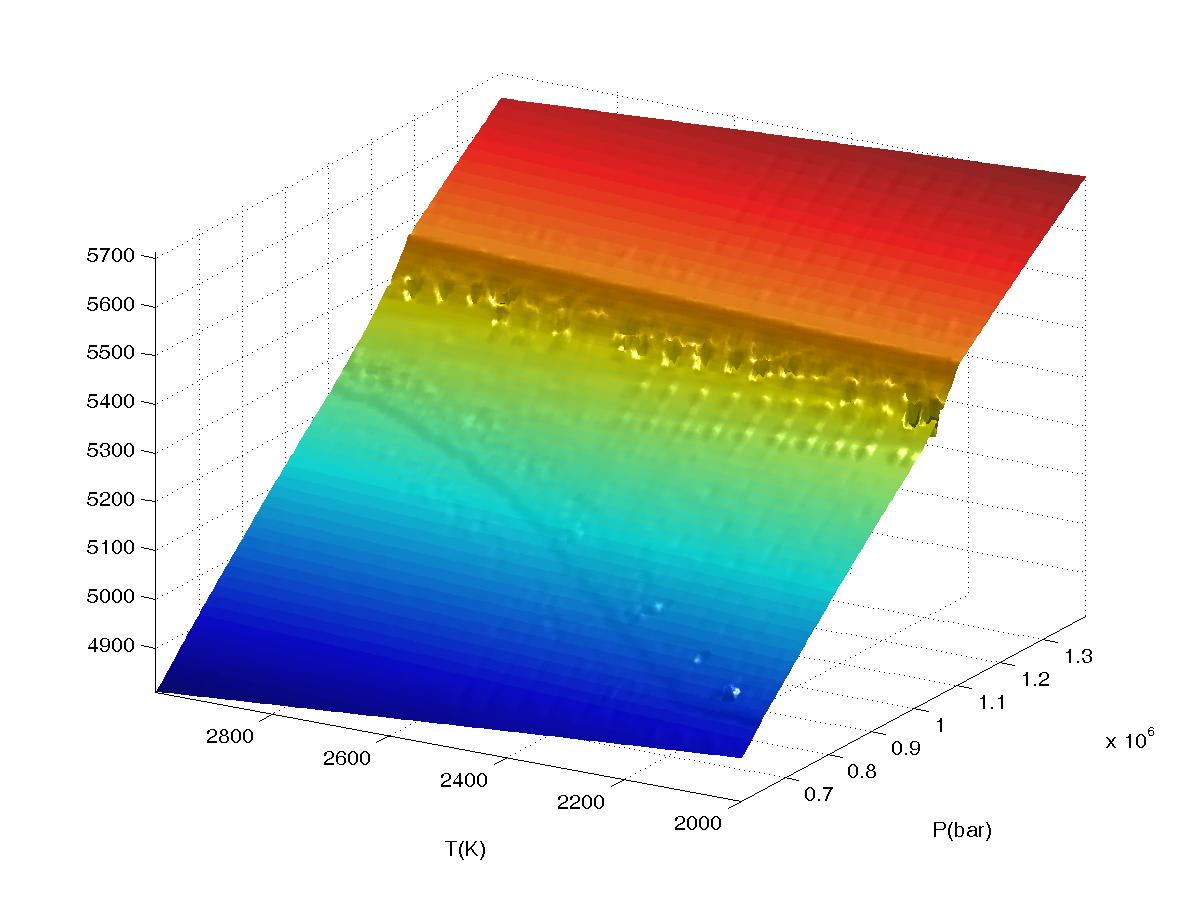
This keyword affects WERAMI.
The
keep_auto keyword controls whether the number of dynamically-generated
compositions retained for each refinement point during adaptive optimization is
automatically reduced if retaining the number of compositions specified by the keep_max
option would exhaust allocated memory (error ver058). Once the threshold
specified by keep_max has been reduced its original value is not restored
during the remainder of a calculation.
If
keep_auto is F and VERTEX or MEEMUM exhausts the memory allocated
for dynamically-generated compositions during an optimization, then the
optimization is reported as failed (warning ver105) and, in the case of VERTEX,
calculations continue.
NOTE:
There is no benefit in setting keep_auto to F unless Perple_X
is recompiled to increase the memory allocated for dynamically generated phase
compositions (parameter k21 in perplex_parameters.h).
This keyword may take values: T (true) or F (false). The default is
T.
This keyword
affects VERTEX and MEEMUM.
keep_max
(obsolete 6.9.1+)
The
keep_max keyword specifies the maximum number of dynamically-generated
compositions retained for each refinement point during adaptive optimization, keep_max
is relevant only if keep_auto is T.
Although
the intent of the keep_max/keep_auto options is primarily to prevent failed
optimizations due to limited memory, in calculations with complex solutions,
reducing keep_max may result in significantly faster calculations with
little reduction in quality. Such effects are problem specific, but in general a
value of 1'000 is adequate, values below 100 may lead to a notable deterioration
in quality and numerical instability.
keep_max
takes integer values > 0, its default is 25000, recommended values are >
100, < k21/10, the value of k21 is specified in perplex_parameters.h and is
generally > 500000.
NOTE:
to maximize computational quality set keep_max to a
large_number (e.g., 5000000). Perple_X
will then reduce keep_max (warning ver058)
as necessary. If keep_max is automatically reduced to a low value (<~20000) it is likely that the problem
is poorly configured or that Perple_X has
been compiled with inappropriate paramters (perplex_parameters.h).
This keyword
affects VERTEX and MEEMUM.
If the linear_model keyword is
on phase boundaries are assumed to be linear between grid nodes at the
active level and next sublevel of a multilevel grid during gridded minimization
calculations. This assumption, which was used
in the original gridded minimization strategy (Connolly 2005), results in a reduction in
the number of calculations because it implies that if the same assemblage is
present at two adjacent nodes at the current level of resolution, then the
assemblage must also be present at the intervening node at the next sublevel.
The assumption is not rigorous and may result in step-like boundaries, a problem
that can be avoided by setting the keyword value to off. When
linear_model is off, the multilevel gridding strategy
is identical to the wavelet strategy described by Gerya et al. (EPSL, 2004). For
2-dimensional grids, setting linear_model to off increases
computation time and output file size roughly twofold.
This keyword affects VERTEX.
The
logarithmic_p keyword controls whether the nominal value of the pressure
variable is interpreted as pressure or its base 10 log. If logarithmic_p is
T, the thermodynamic pressure for calculations is computed as log10P;
thus for a calculation with pressures ranging from 10-5 to 102
bar, the user enters values of -5 and 2 for the minimum and maximum pressure.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword
affects WERAMI, MEEMUM,
and VERTEX.
The
logarithmic_X keyword controls whether the nominal value of the saturated
phase compositional
variable X (usually Y_CO2, the composition of a binary H2O-CO2 fluid) is interpreted as
a composition or its base 10 log. If logarithmic_X is
T, the compositional variable for calculations is computed as log10X;
thus for a calculation with X ranging from 10-5 to 100
mole, the user enters values of -5 and 0 for the minimum and maximum X.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword
affects WERAMI, MEEMUM,
and VERTEX.
The
LP_max_it keyword sets max number of iterations for a single LP
optimization.
This keyword may take
positive integer values. The default is 500.
This keyword
affects MEEMUM
and VERTEX.
NOTE:
low_reach is a not a perplex_option.dat
keyword, the low_reach keyword is an option that applies to a specific
solution model. To implement this keyword modify the solution model file by
adding the keyword to the text of the relevant solution model prior to the end_of_model keyword.
Presence
of the low_reach keyword signals that the composition of a solution is to be
discretized by only two points about a refinement point during the iterative
stage of an adaptive optimization. For a solution with an n-dimensional
composition space this option reduces the usage of memory allocated for dynamic
compositions by a factor of (2/3)^n compared to that required using the default
value of the resolution_factor keyword. For a
10-dimensional solution, this effect reduces memory usage and computation time
by nearly two orders of magnitude. Although these benefits are dramatic, they
are offset by a notable reduction in quality; thus low_reach is an option of
last resort.
low_reach
conflicts with reach_increment, therefore both options should not be set in the
same solution model.
This keyword affects VERTEX and MEEMUM.
See
also: final_resolution, resolution_factor,
initial_resolution, solvus_tolerance, adaptive
minimization keyword group discussion.
The
melt_is_fluid keyword controls whether WERAMI considers silicate melts to
be fluids on output. If melt_is_fluid is F, then properties for
the "solid" aggregate include melts.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects WERAMI and MEEMUM.
The non_linear_switch keyword toggles the default behavior of VERTEX,
MEEMUM, and CONVEX with respect to the discretization parameter XINC used in
non-linear subdivision schemes. See the commnentary
in the header of the 6.8.6+ versions of solution_model.dat for explanation and
discussion of this parameter.
When non_linear_switch is F MEEMUM and VERTEX read XINC from the
text of the individual solution models, otherwise XINC is equated to the
appropriate value of the initial_resolution option.
When non_linear_switch is F CONVEX equates XINC to the appropriate
value of the initial_resolution option, otherwise XINC is read from the text of
the individual solution models.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects VERTEX, MEEMUM, and CONVEX.
The
null_phases keyword controls whether CONVEX considers phases that consist
entirely of mobile and/or saturated components (i.e., a null phase is a phase
that contains no thermodynamic components). The keyword is relevant for
calculations as a function of chemical potentials, activities, or fugacities.
In such calculations phase relations are metastable with respect to null phases
beyond the saturation surface. If null phases are considered, and a
supersaturated initial condition is specified, then CONVEX will terminate with a
message that there are no stable (pseudo-)univariant equilibria. To avoid this
cryptic message it is best to first run such calculations without considering
null phase stability. In chemical potential calculations the saturation surface
can then be computed explicitly by considering null phase stability. In
activity/fugacity calculations the saturation surface cannot be defined
explicitly, but allowing null phases will eliminate metastable equilibria. For
example, if a phase diagram is computed for the CaO-MgO-SiO2 system
as a function of activity of periclase and quartz, then at pressure-temperature
conditions where forsterite is stable the condition a(per) = a(qtz) = 1 is
supersaturated with respect to forsterite; if the same calculation is done as a
function of μ(SiO2) and μ(MgO), then the forsterite
saturation surface will be computed if null phases are allowed.
Null
phases are not permitted in calculations as a function of chemical potentials, activities,
or fugacities with VERTEX. Therefore users should be aware that the phase
equilibria resulting from such calculations may be metastable with respect to
null phases.
This keyword may take values: T (true) or F (false). The default is
F.
option_list_files
The option_list_files controls whether VERTEX, MEEMUM and WERAMI output a
text file that summarizes the option settings relevant for a calculation. If
option_list_files = T, then files are output and named
my_project_PROGRAM_options.txt, where "my_project" is the project name and
PROGRAM is the name of the relevant program.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects VERTEX, MEEMUM and WERAMI.
The isobaric-isothermal Gibbs energy of a solution model with non-convergent
ordering may have several minima as a function of the model order parameters.
These minima correspond to different equilibrium ordering states, the stable
state being that with the lowest Gibbs energy. For such models, Perple_X
is programmed to seek the minima corresponding to the most ordered state. If order_check
is on, the energy of this state is compared to the fully disordered
state, and the lowest energy is used for calculations. Under most conditions,
this strategy
gives the stable state of ordering for all conditions with a single solution
model, but it has the disadvantage that the discontinuous transition between
partially ordered and completely disordered phases (e.g., the tri-critical
curve) is not shown explicitly. If order_check is off, the phase
is always assigned the energy of the maximum order state; therefore in calculations
were an order-disorder model and its fully disordered equivalent are both
present (e.g., omphacite and clinopyroxene), Perple_X
will explicitly delineate discontinuous order-disorder transitions.
If you do not understand the foregoing paragraph, then use the following rules:
1) If order_check = on and the calculation includes an
order-disorder model, then it should NOT include the fully disordered
equivalent (i.e., a calculation including omphacite, should NOT include
clinopyroxene).
2) If order_check is off and the calculation includes an
order-disorder model, then it SHOULD include the fully disordered
equivalent (i.e., a calculation including omphacite, should include
clinopyroxene).
This keyword affects VERTEX, WERAMI, and MEEMUM.
If output_iteration_details is true (T), then MEEMUM and VERTEX write
information on the amount, stability, and composition of refinement points
during each iteration of adaptive
minimization calculations. This output provides diagnostic information on
the quality of minimization results and resolution precision.
The default value of output_iteration_details is F. This keyword is
intended primarily for interactive analysis with MEEMUM, setting output_iteration_details to F
will substantially slow calculations with VERTEX because of the volume of output
written to the console.
See
also: output_iteration_G,
iteration,
initial_resolution, final_resolution,
adaptive
minimization keyword group discussion
If output_iteration_G is true (T), then MEEMUM and VERTEX write
the free energy of the system (per mole of the systems components) and the
chemical potentials of its components after each iteration of adaptive
minimization calculations. This output provides diagnostic information on
the quality of minimization results and resolution precision.
Specifically:
1) The total free energy should converge smoothly to a minimum, increasing the
number of iterations (e.g., by decreasing final_resolution)
beyond this minimum does not improve the quality of the minimization.
2) Oscillation or increasing total free energy during iteration is an indication
of a poor choice of optimization parameters (e.g., over-refinement).
3)
If the free energies of the possible phases of the system are taken to be
perfectly known, then the magnitude of a significant change in the total free
energy of the system is comparable to the round-off error on the reference state
Gibbs energies. Typical round-off error is ~100 J/mol. In general, the true error on
reference state Gibbs energies is ~1000-10000 J/mol.
4)
The precision of minimization may improve significantly even if the improvement
in the total free energy of the solution is insignificant. This precision can be
judged by comparing the variation in the chemical potentials during iteration to
the precision of the data. Chemical potential variations can also be used to
identify well- or poorly-resolved components.
To
illustrate by example, the following output was generated from the bl478_benchmark,
at 1050.15 K and 4000 bar, by increasing final_resolution to 1e-8 mole:
Iteration G(J/mol) H2O MgO Al2O3 K2O CaO TiO2 FeO O2 Na2O SiO2
0 -926202.1570 -390238 -688844 -1794082 -956488 -804320 -1030145 -369515 -512236 -872061 -977953
1 -926270.5179 -391218 -689244 -1794209 -956888 -804224 -1030236 -369536 -511872 -871984 -977953
2 -926284.1030 -390927 -689271 -1794410 -963451 -804091 -1029920 -369108 -525225 -871658 -977953
3 -926288.8551 -391376 -689223 -1794364 -956204 -804244 -1030109 -369072 -522614 -871797 -977953
4 -926289.1760 -391263 -689187 -1794402 -958435 -804194 -1030001 -369150 -522853 -871746 -977953
5 -926289.2590 -391231 -689123 -1794436 -958964 -804152 -1029899 -369190 -523849 -871721 -977953
6 -926289.2753 -391253 -689107 -1794437 -958629 -804150 -1029890 -369180 -524257 -871728 -977953
7 -926289.2797 -391256 -689108 -1794437 -958608 -804151 -1029893 -369179 -524208 -871726 -977953
8 -926289.2811 -391255 -689109 -1794437 -958620 -804152 -1029894 -369179 -524190 -871726 -977953
9 -926289.2816 -391255 -689108 -1794437 -958620 -804151 -1029893 -369179 -524204 -871726 -977953
10 -926289.2817 -391255 -689108 -1794437 -958620 -804152 -1029893 -369179 -524207 -871726 -977953
11 -926289.2818 -391255 -689108 -1794437 -958620 -804152 -1029893 -369179 -524204 -871727 -977953
12 -926289.2818 -391255 -689108 -1794437 -958619 -804152 -1029893 -369179 -524207 -871727 -977953
13 -926289.2818 -391255 -689109 -1794437 -958620 -804152 -1029893 -369179 -524204 -871727 -977953
14 -926289.2818 -391255 -689109 -1794437 -958621 -804152 -1029893 -369179 -524204 -871727 -977953
15 -926289.2818 -391255 -689109 -1794437 -958621 -804152 -1029893 -369179 -524204 -871727 -977953
16 -926289.2818 -391255 -689109 -1794437 -958621 -804152 -1029893 -369179 -524203 -871727 -977953
The
variation in total free energy indicates that the accuracy of the thermodynamic data does not
justify more than 2-3 iterations (as obtained with initial resolution = 0.1 and
final_resolution = 1e-2 mole). The variations in the chemical potentials
indicate that O2 is the most poorly resolved component. Its variation
becomes comparable to its precision after 6 iterations (as obtained with initial
resolution = 0.1 and final_resolution = 1e-3 mole). To put these variations in
perspective, a variation of 50 J/mol in the chemical potential of O2 corresponds
to a variation in oxygen fugacity of 2e-3 log10 units (50 J/mol /(2.303*R*T)).
The default value of output_iteration_G is F. This keyword is
intended primarily for interactive analysis with MEEMUM, setting output_iteration_G to F
will substantially slow calculations with VERTEX because of the volume of output
written to the user console.
See
also: output_iteration_details,
iteration,
initial_resolution, final_resolution,
adaptive
minimization keyword group discussion.
The
pause_on_error (T or F) keyword determines whether Perple_X
programs terminate upon encountering an anticipated error (F) or pause
and wait for user interaction (T).
The default value of pause_on_error is T.
If
a Perple_X program is being run via a
script, or as a subroutine of another program, then pause_on_error should
be set to ensure that in the event of an error Perple_X
returns control to the calling script/program.
In Schreinemakers and mixed-variable diagram calculations (i.e., unconstrained
optimization) compositional degeneracies among the pseudocompounds of different
solutions (e.g., that pseudocompounds of different solutions may have exactly
the same Fe/Mg ratio) may make it impossible for Perple_X
to determine the true variance of pseudo-univariant and -invariant equilibria.
To avoid this problem a small perturbation to the compositions of the
pseudocompounds generated for each solution is introduced. The magnitude of the
perturbation is proportional to the value assigned to the pc_perturbation
keyword.
The default value of pc_perturbation is 5d-3. The sensitivity of
Perple_X to this parameter has not been
tested. The perturbation for each solution is calculated as delta =
pc_perturbation * cst * initial_resolution, where
cst is a random number between 1 and 10. It is expected that this method of
eliminating degeneracies will be effective if delta is greater than the
numerical resolution for compositions (~1d-5).
This keyword affects VERTEX.
The poisson_ratio keyword controls whether, and under what circumstances,
a default value for the Poisson ratio is used to calculate shear moduli and
seismic wave velocities.
The
poisson_ratio
keyword takes 2 values, value1 and value2
value1
may be on, off or all. The default is on
value2 is
the value for the Poisson ratio, the default is 0.35
If value1 = off and no shear modulus is available for a phase, then the
shear modulus and seismic velocities of the phase are assigned the bad_number value.
If value1 = on and no shear modulus is available for a phase,
the shear modulus is computed as 3K(1-2ν)/(ν+1)/2,
where the Poisson ratio ν = value2 and K is the bulk modulus. If no bulk
modulus is available, then the shear modulus and seismic velocities of the phase
are assigned the bad_number value.
If value1 = all, then the shear modulus of any phase is computed as 3K(1-2ν)/(ν+1)/2 regardless of whether other data
are available for the
shear modulus. If no bulk modulus is available, then the shear modulus and
seismic velocities of the phase are assigned the bad_number value.
This option affects WERAMI,
MEEMUM and FRENDLY. For calculations analyzed with WERAMI, changing this keyword
does not necessitate repeating the primary calculation with VERTEX.
Other keywords relevant to seismic
velocity calculations: vrh/hs_weighting,
bounds,
explicit_bulk_modulus, Anderson-Gruneisen, melt_is_fluid,
interpolation, seimsic_data_file.
The
poisson_test (T or F, default F) keyword activates an error trap that
checks whether the poisson_ratio of a phase is negative. The most common cause
of negative Poisson ratios in Perple_X
is a data base configuration in which bulk moduli are computed from an
isostatic EoS while shear moduli are computed from empirical functions.
In
general it is expected that the error trap will only be useful for applications
where seismic wavespeeds are of primary concern. For such applications, running
WERAMI with the error trap activated will identify conditions where the computed
seismic wavespeeds are dubious.
This option affects
WERAMI,
MEEMUM and FRENDLY. For calculations analyzed with WERAMI, changing this keyword
does not necessitate repeating the primary calculation with VERTEX.
The proportions keyword controls whether the relative proportions of
phases (i.e., modes) are determined on the basis of weight or volume.
This keyword may take values: vol, wt. The default is vol.
This option affects WERAMI.
See also: composition_constant, composition_phase,
composition_system
pseudocompound_file
The pseudocompound_file controls whether VERTEX outputs a text file that
summarizes the static pseudocompound compositions used for a calculation. If
pseudocompound_file = T, then the file is output and named
my_project_pseudocompound_list.txt, where my_project is the project name and
PROGRAM is the name of the relevant program.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects VERTEX and MEEMUM.
NOTE:
reach_increment is a not a perplex_option.dat
keyword, the reach_increment keyword is an option that applies to a specific
solution model. To implement this keyword modify the solution model file by
adding the keyword and its value on the line prior to the end_of_model keyword
(e.g., see Figure
4 in the adaptive
minimization keyword group discussion).
The
reach_increment keyword value specifies an incremental increase in the range over which
the compositional variables of a specific solution phase are allowed to vary between successive
iterations during adaptive minimization. The increase may improve the resolution
of phase relations involving two or more solutions with extraordinarily flat
free-energy composition surfaces. This circumstance is problematic, because a small error in the composition of one phase
may cause a large error in the
other phase. Most commonly this occurs when
two or more coexisting phases are related by a solvus. This kind of error is
manifest by spurious solvi, which typically have irregular or patchy boundaries.
This issue is addressed in the adaptive
minimization keyword group discussion.
To
a good approximation, if at iteration n-1 the estimated composition of a
phase is xn-1±dn-1,
then in the subsequent iteration the composition will be allowed to vary
approximately over
the interval xn-1±reach_increment*dn-1.
(see resolution_factor, eq [3]). The
number of compositions considered during the iterative refinement of the
composition of a solution with c compositional variables is proportional to
[1+ resolution_factor + 2*reach_increment]c-1
Thus, large reach_increment values
are particularly expensive in terms of both memory usage and time for complex
solutions (e.g., silicate melts).
The
default value for reach_increment is 0. If reach_increment for a specific
solution model is > global_reach_increment,
then the local value overrides the global value. Experience suggests that a
value of 6 is adequate to resolve phase relations involving silicate
solvi if the initial resolution value for adaptive minimization is
in the range 1/10 - 1/50.
This keyword affects VERTEX and MEEMUM.
See
also: low_reach, final_resolution, resolution_factor,
initial_resolution, solvus_tolerance, adaptive
minimization keyword group discussion.
The
reach_increment_switch keyword controls how reach increments specified by
the
reach_increment and/or global_reach_increment
keywords are implemented.
The
reach_increment_switch keyword may
take the following values:
off
- all reach increments are set to zero
on
- reach increments are applied only during the auto-refine stage of
calculations with VERTEX
all
- reach increments are applied during both the exploratory and auto-refine
stage of calculations with VERTEX
The default is
on.
Setting
reach_increment_switch to all improves the compositional
resolution obtained during the exploratory stage, in general this improvement is
expected to be insignificant and increases both the time and memory required for
the exploratory stage.
For
calculations with MEEMUM reach increments are used regardless of whether
auto-refine data is being used unless reach_increment_switch is set to
off.
This keyword
affects VERTEX and MEEMUM.
The
reaction_format
keyword controls the output format for reactions identified in Schreinemakers
and mixed variable diagram calculations.
This keyword may
take the following values:
minimum
- only phases in the thermodynamic composition space are output
full
- phases in the saturated component composition space are also output
stoich
- stoichiometric coefficients are also output
S+V
- entropy and volume are also output
everything -
everything is output
The default is
minimum.
WARNING: full
reaction equations vary as a function of the independent variables as these
variables may cause the entropy and volume of a reaction and the identity of
stable saturated phases to change. The equations output are those at the first
equilibrium condition found.
This keyword
affects CONVEX.
The
reaction_list
keyword controls whether a spreadsheet formatted list of reactions found during
Schreinemakers and mixed variable diagram calculations.
This keyword may take values:
on, off. The default is off.
This keyword
affects CONVEX.
The
refine_endmembers keyword controls whether the endmember compositions of
a solution are accepted as refinement points during optimization.
This keyword may take
the value T or F.
This keyword
affects VERTEX and MEEMUM.
NOTE: refine_endmembers is not a perplex_option
file
keyword, rather it is an option that applies to a
specific solution model. To implement this keyword modify the solution model by
adding the keyword before the end_of_model keyword.
If
the refine_endmembers keyword is present in a solution model, then the solution model is evaluated to compute the properties of the solution at its
endmember bulk compositions.
Prior
to version 6.7.8, Perple_X did not evaluate
solution models for the composition of a pure endmember, the assumption being
that the properties predicted by
the solution model at the endmember bulk composition would be identical to the
those of the pure endmember. This assumption is not valid
for order-disorder models in which order-disorder is possible at an endmember
bulk composition. In such cases, if refine_endmembers is not specified Perple_X
will return the property of the fully ordered endmember at the endmember
composition, rather than the property of the endmember with the stable state of
order. For example, the Ilm(WPH) model represents order-disorder in terms of an
ordered endmember (oilm) and a disordered endmember (dilm). If no refine_endmembers
keyword is present in the model, Perple_X returns the properties of oilm for the
pure ilmenite bulk composition, but if the refine_endmembers keyword is
added to the model, then the properties obtained for the pure ilmenite bulk composition correspond to an equilibrium mixture of
ordered (oilm) and disordered
ilmenite (dilm) species.
Currently
(6.7.8) there are only two solution
models [Ilm(WPH) and Cpx(G)] in the standard solution model file (solution_model.dat)
where order-disorder occurs in the limit of an endmember bulk composition. The refine_endmembers
keyword has been added to the Ilm(WPH)
model and it must be added to the Cpx(G)
model if Cpx(G) is to be used to compute properties at the bulk Ca-Tschermaks
endmember composition.
The refinement_points keyword specifies the number of metastable, i2,
compositions
that are to be refined during adaptive optimization. Reducing the value of this keyword substantially reduces the cost
(in both memory and time) of calculations, but ultimately may lead to poorly
resolved phase boundaries. Prior to Perple_X
6.7.6, the number of metastable
refinement points was equal to the number of thermodynamic components. From 6.7.6
through 6.8.0 refinement_points defaulted to 5. As of 6.8.1, the default
behavior is equivalent to assigning the aut (automatic)
value.
Unnecessarily large values of this keyword may substantially increase the cost
of calculations in both time and memory, while small values may lead to poorly
resolved phase boundaries and/or numerical instability.
In the initial optimization (iteration 0) of an adaptive optimization
metastable phase compositions are chosen by selecting the least metastable
composition of each solution, subject to the caveat that if a composition of the
solution is stable, the metastable composition must be separated from the stable
composition by a distance > solvus_tolerance_II.
The resulting list of metastable phase compositions is then sorted in order of
increasing metastability and the first i2 compositions of this
list are taken for subsequent refinement. In subsequent iterations, the
metastable refinement points are selected from the compositions associated with
the metastable refinement points of the previous iteration and ranked by
stability.
If
this keyword is assigned the aut (automatic) or default value refinement_points
is set to a minimum value of 5. If c + 2 (c
is the number of thermodynamic components specified for a calculation) > 5,
then refinement_points_II is set to c + 2. If refinement_points
is assigned an integer value in the option file, the specified value is not
modified regardless of the number of thermodynamic components.
This keyword affects VERTEX and MEEMUM.
See
also: low_reach, final_resolution, reach_increment,
initial_resolution, iteration,
solvus_tolerance, adaptive
minimization keyword group discussion, refine_switch.
The
refine_switch keyword controls whether metastable
refinement points
corresponding to a stable phase are retained during the iterative stages of an
adaptive optimization. For problems involving complex solutions, setting this
switch to F may substantially reduce the usage of memory allocated for
dynamic compositions, accelerate calculations, and, counter-intuitively, improve
the resolution of phase boundaries. In simpler problems, setting this switch to T
marginally improves the resolution of phase boundaries.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects VERTEX and MEEMUM.
See
also: low_reach, final_resolution, reach_increment,
initial_resolution, iteration,
solvus_tolerance, adaptive
minimization keyword group discussion,
refinement points.
The
refinement_threshold keyword can be used to specify a threshold in Gibbs energy
beyond which metastable phase compositions will be eliminated during adaptive
optimization. Specifically, if gi - sum(xijmj)
> refinement_threshold, then the composition is eliminated, where mj
are the chemical potentials of the current solution. This may seem
like a good idea. It is not. In systems with a large number of thermodynamic
components, a highly metastable composition may become stable in successive
iterations.
Setting
the refinement threshold to near zero (e.g., 1e-4 J) may result in a several
orders of magnitude reduction in the amount of memory required during adaptive
optimization, but causes only minor reduction in computation time. The fraction
of the total number of compositions eliminated by application of the threshold
is indicated by the console message:
Metastability filtering eliminated XX.XXX% of the trial compositions prior
to optimization.
written
when VERTEX finishes a calculation. Small values may lead to numeric instability
that is manifest by splotchy phase diagram sections and failed optimizations.
The value that qualifies as "small" is problem specific and for that
reason it is not recommended for inexperienced users.
This keyword takes a numeric value (< 12 characters):
>0, and defaults to 1d4. If refinement_threshold > 1d4, then the
threshold is deactivated.
This keyword affects VERTEX and MEEMUM.
See
also: final_resolution, reach_increment,
initial_resolution, iteration,
solvus_tolerance, adaptive
minimization keyword group discussion.
Prior
to version 6.8.4 the iteration keyword took two values, in 6.8.4+ the
second value is specified by refinement_points
and the first value identified as resolution_factor. Eqs [3,4] have been
modified for 6.8.4+ to account for the change in the way reach_increment is
implemented.
In
versions after 6.8.3, but with a time stamp earlier than October 3, 2019,
the range parameter was half the value indicated by Eq [1] below, to obtain
comparable quality to calculations made with those versions with current
versions the refine_factor keyword should be increased to 3.
resolution_factor influences the accuracy with which the compositions of coexisting phases are determined during adaptive
minimization (MEEMUM and VERTEX, Connolly 2009).
In adaptive minimization, the
composition of any solution phase is initially discretized with compositional
resolution d0. and
stable compositions are iteratively refined until the precision is better than the value
of the final_resolution keyword. The initial resolution is not necessarily constant, even for a given
compositional variable in a particular phase (see subdivision_schemes). However,
most commonly, the resolution for all solution models is constant and determined
by initial resolution
keyword (i.e., for Cartesian subdivision). For Cartesian subdivision
d0 = initial resolution.
The specific
resolution formulae given below assume Cartesian subdivision.
In iteration n of an adaptive minimization, the compositional variable of a
phase is allowed to vary discretely over the range ±r around the
estimated composition xn-1. The value of the resolution_factor
keyword,
in combination with d0,
determines both the minimum value of r
rmin =
d0(resolution_factor)2-n/(resolution_factor
+ 1)/2
[1]
and
the actual resolution of the discretization
dn = 2rmin/resolution_factor.
[2]
For a
calculation with a single, miscible, solution phase, at iteration n-1, a composition of
the solution phase xn-1 is determined with precision ±dn-1.
Thus at iteration n, the refined composition xn
will lie within the interval xn-1±dn-1. This simple model is usually
reasonable when multiple solution phases coexist. The exception being in the case that the
free-energy composition surfaces of the phases are extraordinarily flat, as when
two or more coexisting phases are related by a solvus. Solvi are problematic
because a small error in the composition of one phase may cause a large error in the
other immiscible phases. In such circumstances it is helpful to widen the interval over
which a solution may change its composition between iterations. To permit this,
the interval half-width is computed as
rn =
rmin + reach_increment.
[3]
The
number of discretization points for the compositional variable is then
p = 1 + resolution_factor + 2 * reach_increment
[4]
Large values of
resolution_factor increase the time and memory required for
each iteration, but because the rate at which resolution increases with each
iteration also improves with
resolution_factor, fewer
iterations are required to achieve the target precision (final_resolution).
Calculations done with larger values of resolution_factor are slightly more
precise. resolution_factor must be an integer
> 1, its default value is 3, experience suggests that this value is usually optimal.
Changing the value of resolution_factor to resolution_factor' changes the
total number of compositions evaluated by a factor of roughly as (resolution_factor/resolution_factor')c;
for a nine component solution this implies reducing resolution_factor from 3
to 2 will result in a ~38-fold reduction in the total number of compositions
evaluated during an optimization and a comparable reduction in the amount of
memory required for optimizations. If the evaluation of the EoS of the solution
is costly (e.g., in the case of order/disorder models), this reduction
translates directly as a translation in computational time with a relatively minor reduction in the quality of the resulting
calculation. The related reduction in memory is one way of solving the problem indicated
by the VERTEX error message **error
ver058**.
The
resolution_factor keyword takes a real value. It currently defaults to the
minimum permitted value (2), prior to version 6.8.4 the default value of resolution_factor
was 3.
During
adaptive optimization, subdivision using even values of resolution_factor
reproduce the refinement point composition exactly, whereas odd values cause new
compositions to be displaced slightly from the refinement point composition. I
have not tested the consequence of this difference, but my intuition is that
even values should give better results. The factor 2 on reach_increment in [4]
assures reach_increment does not break the even/odd symmetry.
This keyword affects VERTEX and MEEMUM.
See
also: low_reach, final_resolution, reach_increment,
initial_resolution,
refinement_points, output_iteration_G,
solvus_tolerance, adaptive
minimization keyword group discussion.
sample_on_grid
VERTEX
computes phase equilibria and physical properties on multi-level computational
grid. If sample_on_grid is set to T, then WERAMI (in
computational mode 2) samples results on the nodes of the computational grid
used by VERTEX. The advantage of using this option is that it minimizes
interpolation error. Set sample_on_grid to F to sample data on an
arbitrary grid (e.g., to restrict the range of sampling). sample_on_grid has
no effect on the sampling of 1-dimensional grids (WERAMI computational mode 3);
in Perple_X 6.8.5+, 1-dimensional grids are
always sampled on the maximum resolution computational grid used by VERTEX. Note
that only the lowest computational grid used by VERTEX is fully populated by
data; sampling at higher levels normally involves
interpolation.
This keyword may take values: T (true) or F (false). The default is
T.
This keyword affects WERAMI.
The
seismic_data_file
keyword controls whether a file summarizing the options relevant to seismic
wave-speed calculations is output. If requested the file is written to
my_project_seismic_data.txt, where my_project is a place holder for the project
name. The file indicates options specified in perplex_option.dat and provides
lists that indicate how these options are applied to the individual endmembers
and solution models included in a calculation. The file is written whenever
WERAMI, MEEMUM, or VERTEX is executed, therefore caution should be exercised to
ensure that the file is not locked by an editor when these programs are run.
This keyword may take values: T (true) or F (false). The default is
T.
This keyword affects VERTEX, MEEMUM, and WERAMI.
The
seismic_output
keyword controls the amount of information relevant to seismic wave propagation
that is output by the programs WERAMI (in computational mode 1), MEEMUM and
FRENDLY.
This keyword may take values:
all, some, none. The default is some, in which case the
aforementioned programs output wave speeds and adiabatic shear and bulk moduli. If seismic_output = all, then the partial derivatives of wave
speed and elastic moduli with respect to temperature and pressure are also
output. These derivatives do not account for variations in the equilibrium
mineralogy as a function of temperature or pressure.
The short_print
keyword controls whether equilibrium conditions of reactions found during
Schreinemakers and mixed variable diagram calculations are output (provided a
print file is requested).
This keyword may take values:
on, off. The default is on.
This keyword
affects CONVEX.
NOTE: reject_bad_composition is not a perplex_option
file
keyword, rather it is an option that applies to a
specific solution model. To implement this keyword modify the solution model by
adding the keyword before the end_of_model keyword.
...
The
site_check keyword controls whether Perple_X rejects solution model
compositions that yield negative site fractions.
This keyword may take values: T (true) or F (false). The default is
T, i.e., negative site fractions are rejected.
The site populations of a valid solution model must be both positive and a
linear combination of the site populations of its independent endmembers. A
number of solution models implemented by Holland & Powell (JMG 1998, and various
coworkers) in
THERMOCALC, most commonly models involving Tschermaks substitutions, assume
endmembers that are inconsistent with these requirements. To remedy this
inconsistency Holland & Powell impose an ad-hoc correction, referred to as "equipartition",
to the site fractions used to compute configurational entropy. Such ad-hoc corrections are not possible in Perple_X, therefore Perple_X
and THERMOCALC may give different results. These differences can be reduced by
setting
site_check = F (false).
If
site_check = F ,then Perple_X allows solution compositions that
imply negative site fractions. For such compositions the configurational entropy is computed using only the positive site fractions generated by
the invalid model. Thus in this case, Perple_X
allows the same range of solution compositions as THERMOCALC, but does not necessarily compute the same
configurational entropy. If site_check = T, the compositional
range of the solution in Perple_X is
restricted compared to the range allowed in THERMOCALC. With site_check = F results obtained using Perple_X tend to be more
similar to those obtained with THERMOCALC than they are when site_check = T.
Before April 2009, Perple_X operated as though
site_check = F. Therefore Perple_X
results obtained before that date can only be reduced with the current version
if site_check is set explicitly to F.
WARNING: Setting site_check =
F prevents Perple_X from testing for invalid site fractions
that arise for reasons unrelated to the equipartition assumption.
NOTE: site_check_override is not a perplex_option
file
keyword, rather it is an option that applies to a
specific solution model. To implement this keyword modify the solution model by
adding the keyword before the end_of_model keyword.
If
the site_check_override keyword is present in a solution model, then
the tests for invalid dependent endmember site populations introduced in Perple_X
6.7.7 (see Perple_X
6.7.7 for explanation) are overridden and the solution model is treated
in earlier versions of Perple_X.
Specifically, if site_check_override is present in a model, then
dependent endmember properties are computed according to their stoichiometric
definitions in the solution model file and the only constraint on their
concentration in the solution is that no atomic site fraction can be less than
zero.
The
site_check_override is useful for solution models in which
order-disorder effects are partially or entirely accounted for by ad-hoc
assumptions (e.g., Holland & Powell's, 2007, equipartition
assumption).
Currently
(6.7.8) site_check_override has been added to the following solution
models in the standard solution model file (solution_model.dat):
Atg(PN), Amph(DHP), Amph(DPW), Bio(TCC), Chl(HP), GlTrTsPg, GlTrTsMr, Na-Amph.
The
solution_names keyword determines whether Perple_X
uses solution model names, abbreviations, or full_names on output.
This
keyword may take the value mod[el], abb[reviation], or ful[l],
the default is mod. The keyword has no effect for solution models that do
not include the optional abbreviation and full_name keywords.
This keyword affects WERAMI, PSSECT, and MEEMUM.
The solvus_tolerance keyword specifies the
minimum distance (in
normalized composition
space) between two compositions of a solution phase at which the compositions
are considered to represent distinct (immiscible) phases of the solution.
Compositions that are separated by a distance less than solvus_tolerance
are considered to represent a single phase whose composition is computed as the
weighted average of the different compositions of the solution.
This keyword takes the character value aut
(automatic) or numeric values between 0 and 1, it defaults to aut.
solvus_tolerance = aut is almost
always optimal.
Set solvus_tolerance = 0 to prevent VERTEX from
homogenizing stable pseudocompounds that represent a miscible phase (all phase
assemblages will consist of p=c pseudocompounds). Set solvus_tolerance = 1
to make VERTEX homogenize all stable pseudocompounds of a given solution into a
single phase regardless of whether or not the pseudocompounds represent distinct
immiscible compositions.
MEEMUM/VERTEX 6.9.1+:
If solvus_tolerance = aut, then the solvus tolerance is set to 10-2
mol.
CONVEX: If
solvus_tolerance = aut, then the solvus
tolerance is to 3/2 * initial_resolution.
MEEMUM/VERTEX
6.9.0-: For adaptive minimization calculations the most common problems,
i.e., spurious
and stepped solvi, that occur during the calculation of solvus phase
relations can resolved, respectively, via the reach_increment
and solvus_tolerance_II keywords. The default value for solvus_tolerance
(auto), results in a much coarser resolution for adaptive minimization than
specified by final_resolution. The reason this coarse resolution
is used as the default is
that Perple_X may have difficulty
converging to the correct compositions of coexisting immicible phases. These
difficulties (see Figure 2b
of the adaptive
minimization keyword group discussion) arise because the
algorithm assumes that if phase compositions have been discretized with
resolution
dn, then the accuracy
for any compositional variable xn is ±dn.
Therefore during iterative refinement the compositional estimate is not allowed
to move outside the interval xn±dn,
this has the consequence that if two compositions are, in reality, miscible, but
separated by a distance > dn,
the compositional estimates cannot converge during iteration. By improving
the effective initial resolution and/or implementing the reach_increment option
as described in the adaptive
minimization keyword group discussion solvus_tolerance can be reduced to 3/2
* resolution_factor * final_resolution.
This keyword
affects CONVEX, VERTEX, and MEEMUM.
If you do not understand what this keyword does,
then do
not change it.
This parameter can be adjusted to eliminate "stepped"
solvus boundaries along which the compositions of the coexisting phases
change by increments more closely related to the initial_resolution
than to the final_resolution keyword
values.
The solvus_tolerance_II keyword sets an internal
parameter that is used to select metastable phase compositions for subsequent
refinement during adaptive minimization. If in an iteration of an adaptive
minimization calculation, a composition of a solution phase is found to be
stable, then metastable compositions of the solution will only be considered in
the subsequent iteration if the metastable composition is separated from the
stable composition by a distance greater than the value of solvus_tolerance_II.
The idea behind this keyword is that if a solution is immiscible, forcing
Perple_X to consider phases far from a
composition of perceived stability increases the likelihood that the algorithm
will identify the multiple stable phases of a solution. This strategy does seem
to improve the resolution of phase boundaries involving solvi, but for reasons
that are unclear to me it also seems to improve the resolution of phase
boundaries in general. The optimal
value for this keyword should be problem specific, but experience with silicate
solutions seems to suggest that the optimal value for this parameter is
0.25+/-0.05 regardless of the phases under consideration.
A disadvantage of using the relatively large
default value for solvus_tolerance_II is that it may give rise to a numerical
artifact in
Perple_X referred to here as stepped solvi
(See Figure 7 of the adaptive
minimization keyword group discussion for an illustration of a stepped
solvus). This problem occurs when the distance between the limbs
of a solvus are less than the value of solvus_tolerance_II, in this limit
the tolerance prevents the adaptive minimization algorithm from refining
compositions of immiscible phases that are perceived to be metastable. As a
consequence the limbs of the solvus are only located accurately when the
compositions generated at the lowest level of resolution (i.e., as determined by
the effective initial resolution) are
fortuitously close to stable compositions on the limbs of a solvus. The limbs of a solvus
affected by this problem
typically change in abrupt steps that correspond to steps in the compositions of
the immiscible phases that are comparable to the initial_resolution.
An additional disadvantage of setting solvus_tolerance_II to values greater than the effective initial resolution is that it limits the range over
which a stable phase composition may change during adaptive refinement (this limitation can be overcome by using
reach_increment).
Perple_X's adaptive minimization algorithm is such that
it is expected that non-zero values for this keyword improve the resolution of solvi,
but degrade the resolution of normal
phase boundaries by limiting the range over which phases may change their
composition between iterations.
This keyword
affects VERTEX and MEEMUM.
See
also: final_resolution, iteration,
initial_resolution, reach_increment.
The speciation_max_it keyword specifies the maximum number of iterations
permitted for solution of speciation (aka order-disorder) calculations, other
iterative equations of state, and iterative solution of pseudo-univariant
equilibrium conditions (CONVEX).
This keyword takes an integer value (>0) and defaults to 100.
This keyword affects VERTEX, CONVEX, FLUIDS, FRENDLY, WERAMI and MEEMUM.
The
speciation_precision keyword specifies the precision of non-GFSM fluid speciation
calculations.
This keyword takes a numeric value (< 12 characters): <1, and defaults
to 1e-5 mol.
This keyword affects VERTEX, CONVEX, FLUIDS, FRENDLY, WERAMI and MEEMUM.
The
speciation_factor keyword determines the precision of speciation calculations,
this precision is computed as the effective final
resolution divided by the speciation_factor. A value of 100 is usually
adequate. The optimal value of this keyword can be determined by calculating the
total Gibbs energy (with MEEMUM) of a system at a fixed pressure-temperature
condition as a function of the speciation_factor. The smallest value of the
speciation_factor at which no significant variation in the total Gibbs energy
occurs is the optimal value.
Unnecessarily large values of this keyword may substantially slow calculations,
while small values may lead to numerical instability.
This keyword takes a numeric value (< 12 characters):
>10, and defaults to 100.
This keyword affects VERTEX, CONVEX, FLUIDS, FRENDLY, WERAMI and MEEMUM.
This keyword is obsolete in post 6.7.5 versions of Perple_X, see
speciation_factor.
The speciation_tolerance keyword specifies the
precision of speciation calculations for order-disorder models, i.e., the
permitted error in the molar fraction of an ordered species. Prior to Perple_X
version 6.6, the value (10-5) of this tolerance was specified
internally. Typical enthalpies of ordering are in the range 103-104
J/mol; thus a tolerance 10-5 of implies error of <10-1
J/mol in the computed energy of phases described by order-disorder speciation
models. Given that input energies are typically rounded to the nearest 102
J/mol, a speciation_tolerance of 10-2 may be adequate for most
purposes. The advantage of using a large tolerance is that it may accelerate
calculations involving multiple order parameter solution models (e.g., Diener et
al. 2007; Green et al., 2008), the disadvantage of specifying a large tolerance
is that it may increase the roughness of computed properties.
This keyword takes numeric values (< 12 characters):
>0, <1, and defaults to 10-3.
This keyword affects VERTEX, CONVEX, FLUIDS, FRENDLY, WERAMI and MEEMUM.
The species_Gibbs_energies keyword controls whether endmember Gibbs
energies are output by MEEMUM and WERAMI.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword only affects WERAMI and MEEMUM.
The species_output keyword controls whether phase speciation is output by
WERAMI (computational mode 1) and MEEMUM.
This keyword may take values: T (true) or F (false). The default is
T.
This option only affects WERAMI and MEEMUM.
The
spreadsheet keyword controls whether WERAMI includes the independent
variables of a calculation in tabulated output. Setting spreadsheet =
T (true) results in larger output files, but is useful if the data is to be
imported into a spreadsheet program (e.g., Excel).
This keyword may take values: T (true) or F (false). The default is
F.
This keyword
affects mode 2 calculations in WERAMI.
The
description of this option is out-of-date as of the 6.8.1 version of Perple_X,
refer to the header commentary in a 6.8.7+ version of solution_model.dat for a discussion of the non-linear subdivision parameters. The mathematical form
of the stretching transformation given below remains correct, however only IMOD
= 1 is currently supported and the transformation is no longer mapped to [XMIN
XMAX].
The post-3/21/07 version of Perple_X '07
allows four different models for discretizing the composition of a solution
phase. These models, designated "subdivision
schemes" are specified by a line in the solution model
file for
each independently variable endmember consisting of four numbers, XMIN, XMIN,
XINC, and IMOD: XMIN and XMAX are the compositional limits of the solution; the
(initial) number of discrete compositions (i.e., pseudocompounds) is calculated
from XINC (if the initial_resolution keyword
is specified, then the initial_resolution
overrides the value of XINC specified locally in the solution model) as (XMAX-XMIN)/XINC
+ 1; and the spacing of these compositions is determined by IMOD as follows:
IMOD = 0 - Cartesian subdivision, XINC is the (initial) compositional spacing
for the discretization.
IMOD = 1 - asymmetric transform subdivision, compositions are closely
spaced towards XMIN.
IMOD = 2 - symmetric transform subdivision, compositions are closely
spaced towards both XMIN and XMAX.
IMOD = 3 - double symmetric transform subdivision, compositions are closely spaced towards XMIN and XMAX and a third composition that
is
specified on the line following IMOD.
IMOD = 4 - asymmetric transform subdivision, compositions are closely
spaced towards XMAX.
For IMOD=1-4, if the compositional domain is normalized so that W=0 and W=1 represent,
respectively, the compositions at which compositions are closest and furthest apart, compositions
are computed as W =
([1 - Z]*stretch_factor + 2)/(1 + Z) where Z = (stretch_factor +
2)**(1-Y) and Y = 0...1 at constant intervals of dY ~ XINC/(XMAX-XMIN). The exact relation between X and the composition generated depends on
the way W is mapped to X, but to a first approximation, if the stretch_factor
is 0.0164 the resolution of the solution will be improved by an order of
magnitude where the spacing is smallest compared to Cartesian subdivision (IMOD=0).
A consequence of this improvement, is that the resolution of the solution
becomes worse (by more than a factor 2) where the spacing is largest. Increasing
the stretch_factor to values > 1, results in a discretization scheme that is
effectively Cartesian, whereas decreasing the stretch_factor by a factor of 2
increases the maximal compositional
resolution by roughly an order of magnitude. Thus, values <<
0.0164 may destabilize adaptive minimization, while values >> 0.0164 are unlikely
to offer any advantage over the more straightforward Cartesian scheme (IMOD=0).
Pros and cons of non-linear subdivision (IMOD=1-3): The main advantage of
using non-linear subdivision (in particular IMOD=1), is that it permits high
resolution of minor elements (typically Mn, Cr, and Ti) that may strongly
influence the location of phase boundaries.
Calculations of solvi are often sensitive to extremal compositions of a
solution, thus the accuracy of such calculations is improved by specifying IMOD=2
for the relevant solutions, the price paid for this choice is that the
calculation has lower resolution far from the compositional extremes. The major disadvantage of non-linear subdivision is that it has the
potential to generate much larger numbers of compositions (pseudocompounds) for
multicomponent solutions resulting in high costs in terms of computational time
and memory.
The stretch_factor keyword value cannot be < 0, the default value is
0.0164.
This keyword affects VERTEX, CONVEX, and MEEMUM.
The structural_formulae keyword controls whether structural formulae are output
by
WERAMI (in computational mode 1) and MEEMUM.
This keyword may take values: T (true) or F (false). The default is
T.
This option only affects WERAMI and MEEMUM.
The
subdivision_override keyword allows users to override the
subdivision schemes specified locally in the
solution model
file with a single global choice.
subdivision_override may take the value: off, stretch, or
linear. The default is off.
If
subdivision_override = linear, the local values of XMAX, XMIN, and
XINC are interpreted as would be the case if IMOD = 0.
If
subdivision_override = stretch and IMOD = 0, the local values of XMAX,
XMIN, and XINC are interpreted as would be the case if IMOD = 2. If IMOD = 2 or
3 the local values XMAX, XMIN, and XINC are interpreted according to the value
of IMOD.
This keyword affects VERTEX and MEEMUM.
The T_melt keyword specifies a temperature (K) threshold below which the
endmembers of a melt solution model, e.g., melt(HP) and pMELTS(G), are
arbitrarily destabilized. This is sometimes helpful because the thermodynamic
properties of the melt endmembers do not extrapolate to low temperature. This
option only affects melt endmembers that are used by a solution solution model;
thus problematic endmembers that are
present in the data base being used but not included in a model for a stable
melt phase must be excluded explicitly in the problem definition file (the file
created by BUILD). The disadvantage of using T_melt > 0 is that it may
result in an artificial "solidus" at T = T_melt.
The default value
of T_melt is 873.
This keyword affects VERTEX, CONVEX, and MEEMUM.
The T_stop keyword specifies a temperature (K) threshold below which chemical
equilibration is assumed to cease. If the temperature for a calculation is below
the threshold, then the chemical equilibrium is computed at T_stop, but
physical properties for the resulting phase assemblage are computed at the
nominal temperature.
The default value
of T_stop is 0.
This keyword affects VERTEX and MEEMUM.
The timing keyword controls whether VERTEX outputs CPU time usage
statistics. When timing is set to T (true), the output is written to the user
console and a file named my_project.tim, where my_project is the root of the
problem definition file.
This keyword may take values: T (true) or F (false). The default is
T.
This option only affects VERTEX.
NOTE: unbounded_composition is not a perplex_option
file
keyword, rather it is an option that applies to a
specific solution model. To implement this keyword modify the solution model by
adding the keyword before the end_of_model keyword.
If
the unbounded_composition keyword is present in a solution for which the
model composition space is simplicial, then compositions outside the model space
are allowed provided they do not violate site fraction constraints and/or mass
positivity.
This keyword affects VERTEX and MEEMUM.
The variance keyword specifies the maximum true variance of pseudo-univariant
equilibria to be traced during Schreinemakers diagram calculations. Two values
are specified, these are applied, respectively, for the exploratory and
auto-refine stages of calculation.
This keyword affects CONVEX.
vrh/hs_weighting
The value of the vrh/hs_weighting keyword is
the weighting (χ) used to compute aggregate adiabatic elastic moduli for
seismic wave-speed calculations according
to the averaging scheme
Z = (1-χ)*Z- + χ*Z+
where Z- and Z+
are the lower and upper bounds on the aggregate modulus. The method (VRH or HS)
of computing the bounds is specified by the bounds
keyword. For calculations analyzed with WERAMI, changing this keyword does not
necessitate repeating the primary calculation with VERTEX.
This keyword takes numeric values (< 12 characters): 0 -> 1. The default value is 0.5.
This option affects WERAMI,
MEEMUM and FRENDLY.
Other keywords relevant to seismic velocity calculations: bounds,
explicit_bulk_modulus,
poisson_ratio,
Anderson-Gruneisen,
melt_is_fluid,
interpolation, seismic_output, seimsic_data_file.
warn_interactive
VERTEX
and MEEMUM issue intrusive warnings that identify probable bad practice in the
configuration of a problem. After such a warning, the user is offered an
interactive Y/N choice. If the user answers affirmatively, then execution
continues. Setting warning_interactive to F (false) makes these warnings
non-interactive by automatically allowing execution to continue. This setting is NOT
recommended except in instances where the user is certain that the problem
configuration is legitimate.
This keyword may take values: T (true) or F (false). The default is
T.
This keyword affects VERTEX and MEEMUM.
warn_no_limit
To
limit the volume of information written to the console during execution, VERTEX
and MEEMUM limit the number of times certain warnings may be repeated. Setting warn_no_limit
to T (true) overrides these limits for certain warnings.
This keyword may take values: T (true) or F (false). The default is
F.
This keyword affects VERTEX and MEEMUM.
warning_ver637
If immiscible compositions of a solution are stable, then WERAMI automatically
disables
interpolation because of the possibility of interpolating between the wrong
compositions. Setting warning_ver637 to F (false) overrides this
behavior.
For example: if a feldspar solution model is in use, then three
feldspars may coexist. WERAMI distinguishes phase assemblages by the number of
compositions of a solution, but not the compositions; thus, if the order in
which the immiscible phases are identified is different at two nodes of the
computational grid, e.g., feldspar (plagioclase composition) + feldspar (sandine
composition) at one node and feldspar (sanidine composition) + feldspar
(plagioclase composition) at the other, then WERAMI would interpolate incorrectly between the
plagioclase and sandine compositions at the two nodes, resulting in inconsistent
compositions and properties for the feldspar phases. Disabling interpolation
eliminates the possibility of this error, but often results in blocky data
patterns. In general, the ordering of immiscible compositions is consistent between adjacent
nodes and therefore the error is unlikely.
This keyword may take values: T (true) or F (false). The default is
T.
This keyword affects WERAMI.
The zero_bulk keyword specifies the threshold below which a the bulk
composition of a component is considered to be zero. This option is useful
for processing fractionation calculations because in such calculations the
amount of a component may become negligible and destabilize the
calculations. Under these conditions, if zero_bulk is set to a finite value, then
VERTEX will warn of the potential problem. If zero_bulk is assigned a value of zero the option is
effectively off.
This keyword takes numeric values (< 12 characters), 1e-6 is the default
value.
This keyword affects
VERTEX and MEEMUM.
The zero_mode keyword determines whether stable phases present in zero amount
are eliminated from the assemblage on output. This option is particularly useful
for processing fractionation calculations because in such calculations the
amount of one or more components may become zero. The numerical value assigned
to zero_mode defines the tolerance for determining zero amount. If zero_mode is
assigned a negative value the option is turned off.
This keyword takes numeric values (< 12 characters): 0 -> 1, 1e-6 is the default
value except for fractionation calculations. For fractionation calculations
zero_mode defaults (is fixed?) to zero.
This keyword affects
VERTEX and MEEMUM.
1d_path
For phase fractionation calculations the 1d_path keyword indicates
the number of nodes taken to define the path in the exploratory and auto-refine
stages of the calculation. The default values for 1d_path are 20 and 150 for the
exploratory and auto-refinement stages of a phase fractionation calculation. As
a rule of thumb, the latter value should be chosen such that the results do not
change significantly if it is varied by a factor of two.
This keyword affects VERTEX.